I am excited to share that our paper XTrace: Making the most of every sample in stochastic trace estimation has been published in the SIAM Journal on Matrix Analysis and Applications. (See also our paper on arXiv.)
Spurred by this exciting news, I wanted to take the opportunity to share one of my favorite results in randomized numerical linear algebra: a “speed limit” result of Meyer, Musco, Musco, and Woodruff that establishes a fundamental limitation on how accurate any trace estimation algorithm can be.
Let’s back up. Given an unknown square matrix  , the trace of , defined to be the sum of its diagonal entries
, the trace of , defined to be the sum of its diagonal entries
![\[\tr(A) \coloneqq \sum_{i=1}^n A_{ii}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-80cd9a9ca29d1d3bb13ce28fd80b5e59_l3.png "Rendered by QuickLaTeX.com")
through matrix–vector products (affectionately known as “matvecs”): Given any vector  , we have access to
, we have access to  . Our goal is to form an estimate
. Our goal is to form an estimate  that is as accurate as possible while using as few matvecs as we can get away with.
that is as accurate as possible while using as few matvecs as we can get away with.
To simplify things, let’s assume the matrix is symmetric and positive (semi)definite. The classical algorithm for trace estimation is due to Girard and Hutchinson, producing a probabilistic estimate with a small average (relative) error:
![\[\expect\left[\frac{|\hat{\tr}-\tr(A)|}{\tr(A)}\right] \le \varepsilon \quad \text{using } m= \frac{\rm const}{\varepsilon^2} \text{ matvecs}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-07898a8a2214416cb97b8b72956e3e76_l3.png "Rendered by QuickLaTeX.com")
 ) requires roughly
) requires roughly  matvecs!
matvecs!
This state of affairs was greatly improved by Meyer, Musco, Musco, and Woodruff. Building upon previous work, they proposed the Hutch++ algorithm and proved it outputs an estimate satisfying the following bound:
(1) ![\[\expect\left[\frac{|\hat{\tr}-\tr(A)|}{\tr(A)}\right] \le \varepsilon \quad \text{using } m= \frac{\rm const}{\varepsilon} \text{ matvecs}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b9f186640222c78e5064f83465038b78_l3.png "Rendered by QuickLaTeX.com")
 matvecs to achieve 1% error! Our algorithm, XTrace, satisfies the same error guarantee (1) as Hutch++. On certain problems, XTrace can be quite a bit more accurate than Hutch++.
matvecs to achieve 1% error! Our algorithm, XTrace, satisfies the same error guarantee (1) as Hutch++. On certain problems, XTrace can be quite a bit more accurate than Hutch++.
The MMMW Trace Estimation “Speed Limit”
Given the dramatic improvement of Hutch++ and XTrace over Girard–Hutchinson, it is natural to hope: Is there an algorithm that does even better than Hutch++ and XTrace? For instance, is there an algorithm satisfying an even slightly better error bound of the form
![\[\expect\left[\frac{|\hat{\tr}-\tr(A)|}{\tr(A)}\right] \le \varepsilon \quad \text{using } m= \frac{\rm const}{\varepsilon^{0.999}} \text{ matvecs}?\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8bae682300a401dc80869764b302ce8c_l3.png "Rendered by QuickLaTeX.com")
Let’s add some fine print. Consider an algorithm for the trace estimation problem. Whenever the algorithm wants, it can present a vector  and receive back
and receive back  . The algorithm is allowed to be adaptive: It can use the matvecs
. The algorithm is allowed to be adaptive: It can use the matvecs  it has already collected to decide which vector
it has already collected to decide which vector  to present next. We measure the cost of the algorithm in terms of the number of matvecs alone, and the algorithm knows nothing about the psd matrix other what it learns from matvecs.
to present next. We measure the cost of the algorithm in terms of the number of matvecs alone, and the algorithm knows nothing about the psd matrix other what it learns from matvecs.
One final stipulation:
Simple entries assumption. We assume that the entries of the vectors
and
with up to
digits after the decimal place.
To get a feel for this simple entries assumption, suppose we set  . Then
. Then  would be an allowed input vector, but
would be an allowed input vector, but  would not be (too many digits after the decimal place). Similarly,
would not be (too many digits after the decimal place). Similarly,  would not be valid because its entries exceed . The simple entries assumption is reasonable as we typically represent numbers on digital computers by storing a fixed number of digits of accuracy.1We typically represent numbers on digital computers by floating point numbers, which essentially represent numbers using scientific notation like
would not be valid because its entries exceed . The simple entries assumption is reasonable as we typically represent numbers on digital computers by storing a fixed number of digits of accuracy.1We typically represent numbers on digital computers by floating point numbers, which essentially represent numbers using scientific notation like  . For this analysis of trace estimation, we use fixed point numbers like
. For this analysis of trace estimation, we use fixed point numbers like  (no powers of ten allowed)!
(no powers of ten allowed)!
With all these stipulations, we are ready to state the “speed limit” for trace estimation proved by Meyer, Musco, Musco, and Woodruff:
Informal theorem (Meyer, Musco, Musco, Woodruff). Under the assumptions above, there is no trace estimation algorithm producing an estimate
![\[\expect\left[\frac{|\hat{\tr}-\tr(A)|}{\tr(A)}\right] \le \varepsilon \quad \text{using } m= \frac{\rm const}{\varepsilon^{0.999}} \text{ matvecs}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9034edba9bafb68809d24d54f5a830e6_l3.png "Rendered by QuickLaTeX.com")
We will see a slightly sharper version of the theorem below, but this statement captures the essence of the result.
Communication Complexity
To prove the MMMW theorem, we have to take a journey to the beautiful subject of communication complexity. The story is this. Alice and Bob are interested in solving a computational problem together. Alice has her input and Bob has his input  , and they are interested in computing a function
, and they are interested in computing a function  of both their inputs.
of both their inputs.
Unfortunately for the two of them, Alice and Bob are separated by a great distance, and can only communicate by sending single bits (0 or 1) of information over a slow network connection. Every bit of communication is costly. The field of communication complexity is dedicated to determining how efficiently Alice and Bob are able to solve problems of this form.
The Gap-Hamming problem is one example of a problem studied in communication complexity. As inputs, Alice and Bob receive vectors  with
with  and entries from a third party Eve. Eve promises Alice and Bob that their vectors and satisfy one of two conditions:
and entries from a third party Eve. Eve promises Alice and Bob that their vectors and satisfy one of two conditions:
(2) ![\[\text{Case 0: } x^\top y \ge\sqrt{n} \quad \text{or} \quad \text{Case 1: } x^\top y \le -\sqrt{n}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3134941eeedf133f83ba4795b499332f_l3.png "Rendered by QuickLaTeX.com")
There’s one simple solution to this problem: First, Bob sends his whole input vector to Alice. Each entry of takes one of the two value  and can therefore be communicated in a single bit. Having received , Alice computes
and can therefore be communicated in a single bit. Having received , Alice computes  , determines whether they are in case 0 or case 1, and sends Bob a single bit to communicate the answer. This procedure requires
, determines whether they are in case 0 or case 1, and sends Bob a single bit to communicate the answer. This procedure requires  bits of communication.
bits of communication.
Can Alice and Bob still solve this problem with many fewer than  bits of communication, say
bits of communication, say  bits? Unfortunately not. The following theorem of Chakrabati and Regev shows that roughly bits of communication are needed to solve this problem:
bits? Unfortunately not. The following theorem of Chakrabati and Regev shows that roughly bits of communication are needed to solve this problem:
Theorem (Chakrabati–Regev). Any algorithm which solves the Gap-Hamming problem that succeeds with at least
probability for every pair of inputs
bits of communication.
Here, is big-Omega notation, closely related to big-O notation  and big-Theta notation
and big-Theta notation  . For the less familiar, it can be helpful to interpret , , and as all standing for “proportional to ”. In plain language, the theorem of Chakrabati and Regev result states that there is no algorithm for the Gap-Hamming problem that much more effective than the basic algorithm where Bob sends his whole input to Alice (in the sense of requiring less than bits of communication).
. For the less familiar, it can be helpful to interpret , , and as all standing for “proportional to ”. In plain language, the theorem of Chakrabati and Regev result states that there is no algorithm for the Gap-Hamming problem that much more effective than the basic algorithm where Bob sends his whole input to Alice (in the sense of requiring less than bits of communication).
Reducing Gap-Hamming to Trace Estimation
This whole state of affairs is very sad for Alice and Bob, but what does it have to do with trace estimation? Remarkably, we can use hardness of the Gap-Hamming problem to show there’s no algorithm that fundamentally improves on Hutch++ and XTrace. The argument goes something like this:
- If there were a trace estimation algorithm fundamentally better than Hutch++ and XTrace, we could use it to solve Gap-Hamming in fewer than bits of communication.
- But no algorithm can solve Gap-Hamming in fewer than bits or communication.
- Therefore, no trace estimation algorithm is fundamentally better than Hutch++ and XTrace.
Step 2 is the work of Chakrabati and Regev, and step 3 follows logically from 1 and 2. Therefore, we are left to complete step 1 of the argument.
Protocol
Assume we have access to a really good trace estimation algorithm. We will use it to solve the Gap-Hamming problem. For simplicity, assume is a perfect square. The basic idea is this:
- Have Alice and Bob reshape their inputs into matrices
 , and consider (but do not form!) the positive semidefinite matrix
, and consider (but do not form!) the positive semidefinite matrix ![\[A = (X+Y)^\top (X+Y).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2b1df104e3d571430963573b54d5c4df_l3.png "Rendered by QuickLaTeX.com")
- Observe that
Thus, the two cases in (2) can be equivalently written in terms of![\[\tr(A) = \tr(X^\top X) + 2\tr(X^\top Y) + \tr(Y^\top Y) = 2n + 2(x^\top y).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a0f6e916740a9c27733c659418cc15cf_l3.png "Rendered by QuickLaTeX.com")
 :
:(2′)
![\[\text{Case 0: } \tr(A)\ge 2n + 2\sqrt{n} \quad \text{or} \quad \text{Case 1: } \tr(A) \le 2n-2\sqrt{n}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1fea970636b48c827e4584d3655b9e3c_l3.png "Rendered by QuickLaTeX.com")
- By working together, Alice and Bob can implement a trace estimation algorithm. Alice will be in charge of running the algorithm, but Alice and Bob must work together to compute matvecs. (Details below!)
- Using the output of the trace estimation algorithm, Alice determines whether they are in case 0 or 1 (i.e., where
 or
or  ) and sends the result to Bob.
) and sends the result to Bob.
To complete this procedure, we just need to show how Alice and Bob can implement the matvec procedure using minimal communication. Suppose Alice and Bob want to compute  for some vector
for some vector  with entries between and with up to decimal digits. First, convert to a vector
with entries between and with up to decimal digits. First, convert to a vector  whose entries are integers between
whose entries are integers between  and
and  . Since
. Since  , interconverting between and
, interconverting between and  is trivial. Alice and Bob’s procedure for computing is as follows:
is trivial. Alice and Bob’s procedure for computing is as follows:
- Alice sends Bob
 .
. - Having received , Bob forms
 and sends it to Alice.
and sends it to Alice. - Having received , Alice computes
 and sends it to Bob.
and sends it to Bob. - Having received
 , Bob computes
, Bob computes  and sends its to Alice.
and sends its to Alice. - Alice forms
 .
.
Because  and
and  are
are  and have entries, all vectors computed in this procedure are vectors of length with integer entries between
and have entries, all vectors computed in this procedure are vectors of length with integer entries between  and
and  . We conclude the communication cost for one matvec is
. We conclude the communication cost for one matvec is  bits.
bits.
Analysis
Consider an algorithm we’ll call BestTraceAlgorithm. Given any accuracy parameter  , BestTraceAlgorithm requires at most
, BestTraceAlgorithm requires at most  matvecs and, for any positive semidefinite input matrix of any size, produces an estimate satisfying
matvecs and, for any positive semidefinite input matrix of any size, produces an estimate satisfying
(3) ![\[\expect\left[\frac{|\hat{\tr}-\tr(A)|}{\tr(A)}\right] \le \varepsilon.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a9165ea3846d540eb654af9f7f59a5c2_l3.png "Rendered by QuickLaTeX.com")
 matvecs.
matvecs.
To solve the Gap-Hamming problem, Alice and Bob just need enough accuracy in their trace estimation to distinguish between cases 0 and 1. In particular, if
![\[\left| \frac{\hat{\tr} - \tr(A)}{\tr(A)} \right| \le \frac{1}{\sqrt{n}},\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8327e1b03c9ba9162fb737e40a192b3a_l3.png "Rendered by QuickLaTeX.com")
Suppose that Alice and Bob apply trace estimation to solve the Gap-Hamming problem, using  matvecs in total. The total communication is
matvecs in total. The total communication is  bits. Chakrabati and Regev showed that Gap-Hamming requires
bits. Chakrabati and Regev showed that Gap-Hamming requires  bits of communication (for some
bits of communication (for some  ) to solve the Gap-Hamming problem with probability. Thus, if
) to solve the Gap-Hamming problem with probability. Thus, if  , then Alice and Bob fail to solve the Gap-Hamming problem with at least
, then Alice and Bob fail to solve the Gap-Hamming problem with at least  probability. Thus,
probability. Thus,
![\[\text{If } m < \frac{cn}{T} = \Theta\left( \frac{\sqrt{n}}{b+\log n} \right), \quad \text{then } \left| \frac{\hat{\tr} - \tr(A)}{\tr(A)} \right| > \frac{1}{\sqrt{n}} \text{ with probability at least } \frac{1}{3}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-74364fd915239fdaf0889d005378b7ba_l3.png "Rendered by QuickLaTeX.com")
![\[\text{If }\left| \frac{\hat{\tr} - \tr(A)}{\tr(A)} \right| \le \frac{1}{\sqrt{n}}\text{ with probability at least } \frac{2}{3}, \quad \text{then } m \ge \Theta\left( \frac{\sqrt{n}}{b+\log n} \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-81fbd6e8e08c9759a6cbf5ac4f9782e2_l3.png "Rendered by QuickLaTeX.com")
Say Alice and Bob run BestTraceAlgorithm with parameter
 . Then, by (3) and Markov’s inequality,
. Then, by (3) and Markov’s inequality, ![\[\left| \frac{\hat{\tr} - \tr(A)}{\tr(A)} \right| \le \frac{1}{\sqrt{n}} \quad \text{with probability at least }\frac{2}{3}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-95db791999f5573659f14f5b0d4a386e_l3.png "Rendered by QuickLaTeX.com")
![\[m \ge \Theta\left( \frac{\sqrt{n}}{b+\log n} \right) \text{ matvecs}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8f0975698051029d6273269409146a9f_l3.png "Rendered by QuickLaTeX.com")
 , we conclude that any trace estimation algorithm, even BestTraceAlgorithm, requires
, we conclude that any trace estimation algorithm, even BestTraceAlgorithm, requires ![\[m \ge \Theta \left( \frac{1}{\varepsilon (b+\log(1/\varepsilon))} \right) \text{ matvecs}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b3eb651ca114a7fe19f3b8c5d3e64063_l3.png "Rendered by QuickLaTeX.com")
 using even
using even  matvecs. This proves the MMMW theorem.
matvecs. This proves the MMMW theorem.
 be a domain and let
be a domain and let  be a (
be a ( . We consider the task of evaluating
. We consider the task of evaluating ![\[I[f] = \int_\Omega f(x) g(x) \, \mathrm{d}\mu(x).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4767401252e7558c87b5a518f3609468_l3.png "Rendered by QuickLaTeX.com")
 ,
, ![I[f]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-603f22dedcf932b6f47e37160c09afd8_l3.png "Rendered by QuickLaTeX.com") for multiple different functions
for multiple different functions  .
.
![\[\hat{I}_{w,s}[f] = \sum_{i=1}^n w_i f(s_i) \approx I[f].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4bb6a83827b630719dd50338a790b9c6_l3.png "Rendered by QuickLaTeX.com")
 and points
and points  such that the approximation
such that the approximation ![\hat{I}_{w,s}[f] \approx I[f]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-61580a8cc2043483ce9c1ab09e3ea85e_l3.png "Rendered by QuickLaTeX.com") is accurate.
is accurate.
 be an RKHS with
be an RKHS with  . We can interpret the norm as assigning a roughness
. We can interpret the norm as assigning a roughness  to each function
to each function  . The kernel is a bivariate function
. The kernel is a bivariate function  . It is related to the RKHS
. It is related to the RKHS  by the reproducing property
by the reproducing property![\[f(x)=\langle f, k(x,\cdot)\rangle \quad \text{for every }f\in\mathcal{H},x\in\Omega.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-276db3513a1ea34da49f342ad7f4a7ea_l3.png "Rendered by QuickLaTeX.com")
 represents the univariate function obtained by setting the first input of
represents the univariate function obtained by setting the first input of  . Let’s first assume that the nodes
. Let’s first assume that the nodes  are fixed, and talk about how to pick the weights
are fixed, and talk about how to pick the weights  that we’ll called the ideal weights. There (at least) are five equivalent ways of characterizing the ideal weights. We’ll present all of them. As an exercise, you can try and convince yourself that these characterizations are equivalent, giving rise to the same weights.
that we’ll called the ideal weights. There (at least) are five equivalent ways of characterizing the ideal weights. We’ll present all of them. As an exercise, you can try and convince yourself that these characterizations are equivalent, giving rise to the same weights. .
.![\[\hat{I}_{w_\star,s}[k(s_i,\cdot)]=I[k(s_i,\cdot)] \quad \text{for } i=1,2,\ldots,n.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-52bf51e5f4ddb42195d5896ff71ebe02_l3.png "Rendered by QuickLaTeX.com")
 :
:![\[\sum_{j=1}^n k(s_i,s_j)w^\star_j = \int_\Omega k(s_i,x) g(x)\,\mathrm{d}\mu(x) \quad \text{for }i=1,2,\ldots,n.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1a5ca36966f56dae9d994e208f9ef036_l3.png "Rendered by QuickLaTeX.com")
 is the ideal weights.
is the ideal weights.
 at the nodes, obtaining an interpolant
at the nodes, obtaining an interpolant  . Then, obtain an approximation to the integral by integrating the interpolant:
. Then, obtain an approximation to the integral by integrating the interpolant:![\[\hat{I}_{w^\star,s}[f] \coloneqq \int_\Omega \hat{f}(x) g(x) \, \mathrm{d}\mu(x).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0c68060fdf085f41d9cbdd9cc5c16e31_l3.png "Rendered by QuickLaTeX.com")
![\[\hat{f} = \argmin \{ \norm{h} : h(s_i) = f(s_i) \text{ for } i=1,\ldots,n\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5619a986180c554d2a079e5156a97571_l3.png "Rendered by QuickLaTeX.com")
![\[\hat{f} = \sum_{i=1}^n \alpha_i k(\cdot,s_i)\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0abcded0eda92deac99b904817afdf5b_l3.png "Rendered by QuickLaTeX.com")
 .With a little algebra, you can show that the integral of
.With a little algebra, you can show that the integral of ![\[I[\hat{f}] = \sum_{i=1}^n w^\star_i f(s_i),\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f0da23bc7dc97894b0a6702b6bf6ddfb_l3.png "Rendered by QuickLaTeX.com")
![\[\operatorname{Err}(w,s)=\sup_{\norm{f}\le 1}\left| I[f] - \hat{I}_{w,s}[f]\right|.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6e46307dc4cdac54a3ab010b36ed7858_l3.png "Rendered by QuickLaTeX.com")
 is the highest possible quadrature error for a function
is the highest possible quadrature error for a function  of norm at most 1.
of norm at most 1.
![\[w^\star=\operatorname*{argmin}_{w\in\real^n}\operatorname{Err}(w,s).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-06e68886b8279d9d592cae3775d4295f_l3.png "Rendered by QuickLaTeX.com")
 for a mean-zero Gaussian process with
for a mean-zero Gaussian process with ![\[\Cov(f(x),f(y))=k(x,y)\quad \text{for every } x,y\in\Omega.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-19fea8ea8406c891c0d6bab2166dc4a4_l3.png "Rendered by QuickLaTeX.com")
![\[\operatorname{MSE}(w,s)\coloneqq \expect_{f\sim\operatorname{GP}(0,k)} \left( I[f] - \hat{I}_{w,s}[f] \right)^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4741e0bcba7276b0a3a9266cd61b54c0_l3.png "Rendered by QuickLaTeX.com")
![\[\operatorname{MSE}(w,s)=\operatorname{Err}(w,s)^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-dc387f1211b9e5a070f2778a8205c022_l3.png "Rendered by QuickLaTeX.com")
![\[w^\star=\operatorname*{argmin}_{w\in\real^n}\operatorname{MSE}(w,s).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-32506adbc7176da52a920dbf4c373149_l3.png "Rendered by QuickLaTeX.com")
 . The integral of this random function
. The integral of this random function ![I[h]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e47eba0764d69669e85367cf245d71d3_l3.png "Rendered by QuickLaTeX.com") is a random variable. To numerically integrate a function
is a random variable. To numerically integrate a function  agreeing with
agreeing with ![\[\hat{I}_{w^\star,s}[f]\coloneqq \expect_{h\sim\operatorname{GP}(0,k)}[I[h] \mid h(s_i)=f(s_i) \text{ for } i=1,\ldots,n].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ce1156fb9da612607ae0158770a34645_l3.png "Rendered by QuickLaTeX.com")
 , it is reasonable to add an additional constraint that the weights
, it is reasonable to add an additional constraint that the weights ![\[w\in\Delta\coloneqq \left\{ p\in\real^n_+ : \sum_{i=1}^n p_i = 1\right\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-55c593b2593abc9ea8c18d4f8edbccd3_l3.png "Rendered by QuickLaTeX.com")
 ; thus, in effect, quadrature amounts to approximating one probability measure
; thus, in effect, quadrature amounts to approximating one probability measure  . Additional constraints such as these can easily be imposed when using the optimization characterizations 3 and 4 of the ideal weights. See
. Additional constraints such as these can easily be imposed when using the optimization characterizations 3 and 4 of the ideal weights. See  ? To pick the nodes, it seems sensible to try and minimize the worst-case error
? To pick the nodes, it seems sensible to try and minimize the worst-case error  with the ideal weights
with the ideal weights ![\[\operatorname{Err}(w^\star,s) = \norm{\int_\Omega (k(\cdot,x) - \hat{k}_s(\cdot,x)) g(x) \, \mathrm{d}\mu(x)}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f66a0cdfc8a943a944dad0fed98dbad9_l3.png "Rendered by QuickLaTeX.com")
 is the
is the ![\[\hat{k}_s(x,y) = k(x,s) k(s,s)^{-1} k(s,y).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9c55d13be5a7cdb27139303cb08ec19f_l3.png "Rendered by QuickLaTeX.com")
 for the kernel matrix with
for the kernel matrix with  entry
entry  and
and  and
and  for the row and column vectors with
for the row and column vectors with  th entry
th entry  and
and  .
.
 as accurate as possible.
as accurate as possible. is and, thus, the smaller the error
is and, thus, the smaller the error  , it has a high probability of placing the next node
, it has a high probability of placing the next node  far from the previously selected nodes.
far from the previously selected nodes.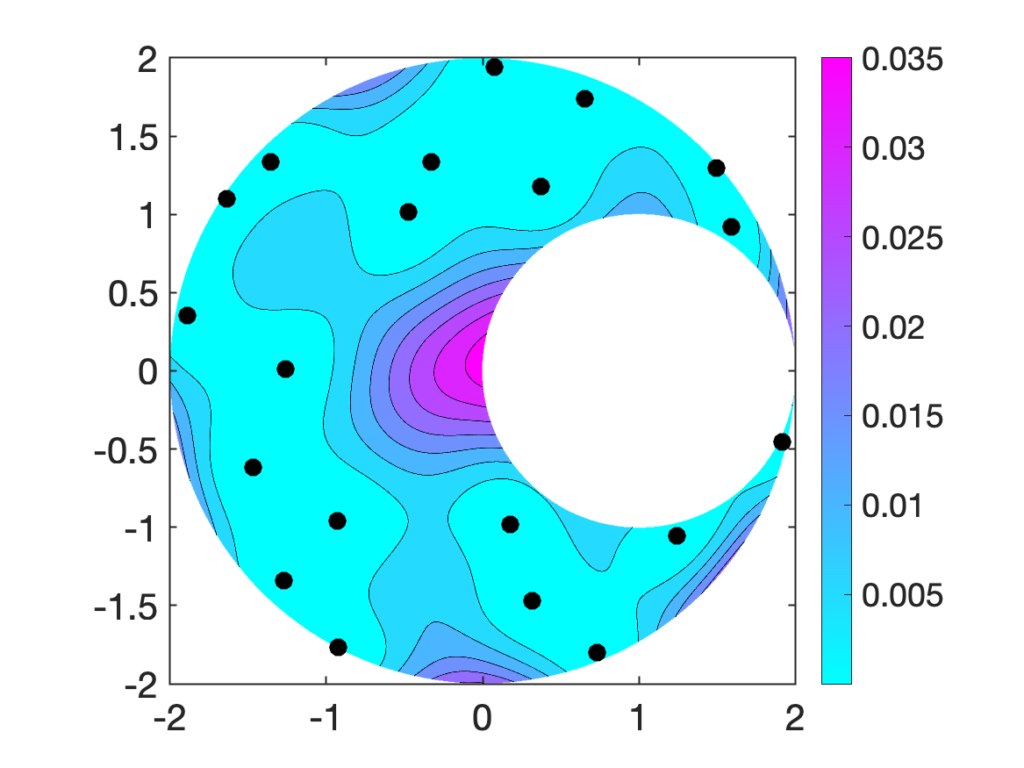
 seeks to compress a high-dimensional matrix
seeks to compress a high-dimensional matrix  or vector
or vector  to a lower-dimensional sketched matrix
to a lower-dimensional sketched matrix  or vector
or vector  . The quality of a sketching matrix for a matrix
. The quality of a sketching matrix for a matrix ![\[(1-\varepsilon) \norm{x} \le \norm{Sx} \le (1+\varepsilon) \norm{x} \quad \text{for every } x \in \operatorname{col}(A).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-06811a2d05bdccddd7b3c3079ed47729_l3.png "Rendered by QuickLaTeX.com")
 denotes the
denotes the  .
. matrix.
matrix. (one million) and output dimension
(one million) and output dimension  . For the SRTT, we use the
. For the SRTT, we use the  .
.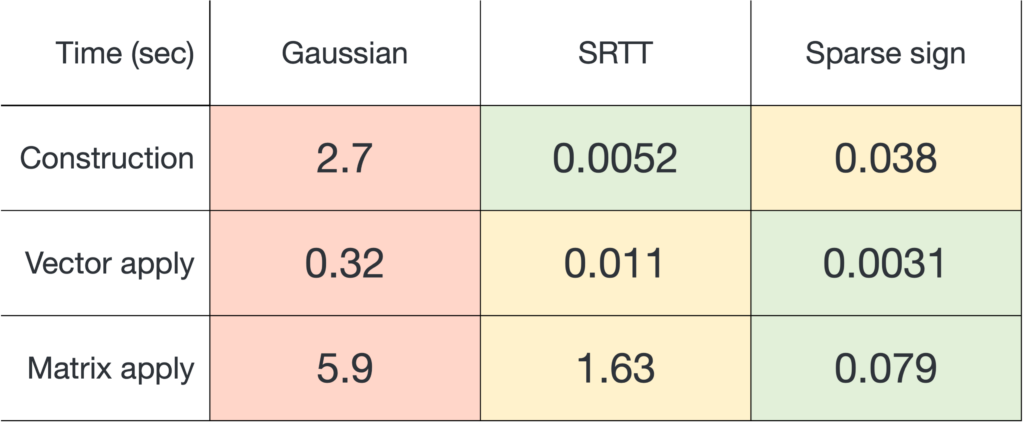
 and applying it to a matrix
and applying it to a matrix  , sparse sign embeddings are 14× faster than SRTTs and 73× faster than Gaussian embeddings.
, sparse sign embeddings are 14× faster than SRTTs and 73× faster than Gaussian embeddings. for
for  and
and  using the MATLAB
using the MATLAB  between 100 and 10,000. We report the distortion averaged over 100 trials. The theoretically predicted value
between 100 and 10,000. We report the distortion averaged over 100 trials. The theoretically predicted value  (equivalently,
(equivalently,  ) is shown as a dashed line.
) is shown as a dashed line.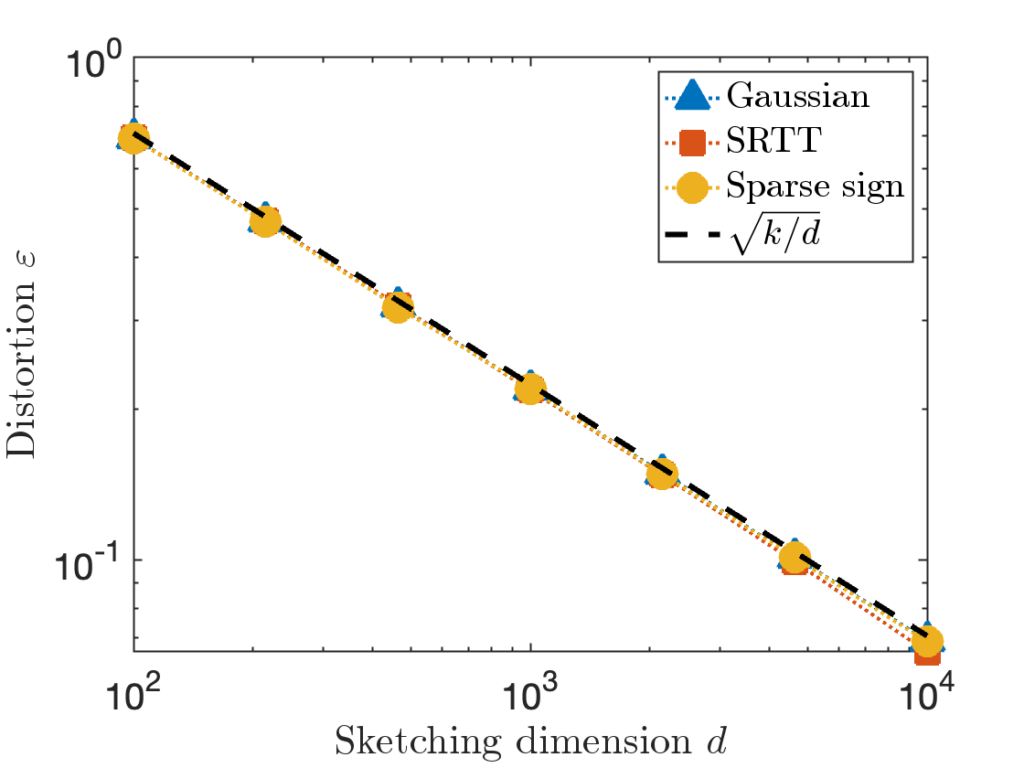
 is taken to be a matrix with independent standard Gaussian random values.
is taken to be a matrix with independent standard Gaussian random values. is taken to be the
is taken to be the  is taken to be the
is taken to be the  identity matrix stacked onto a
identity matrix stacked onto a  matrix of zeros.
matrix of zeros.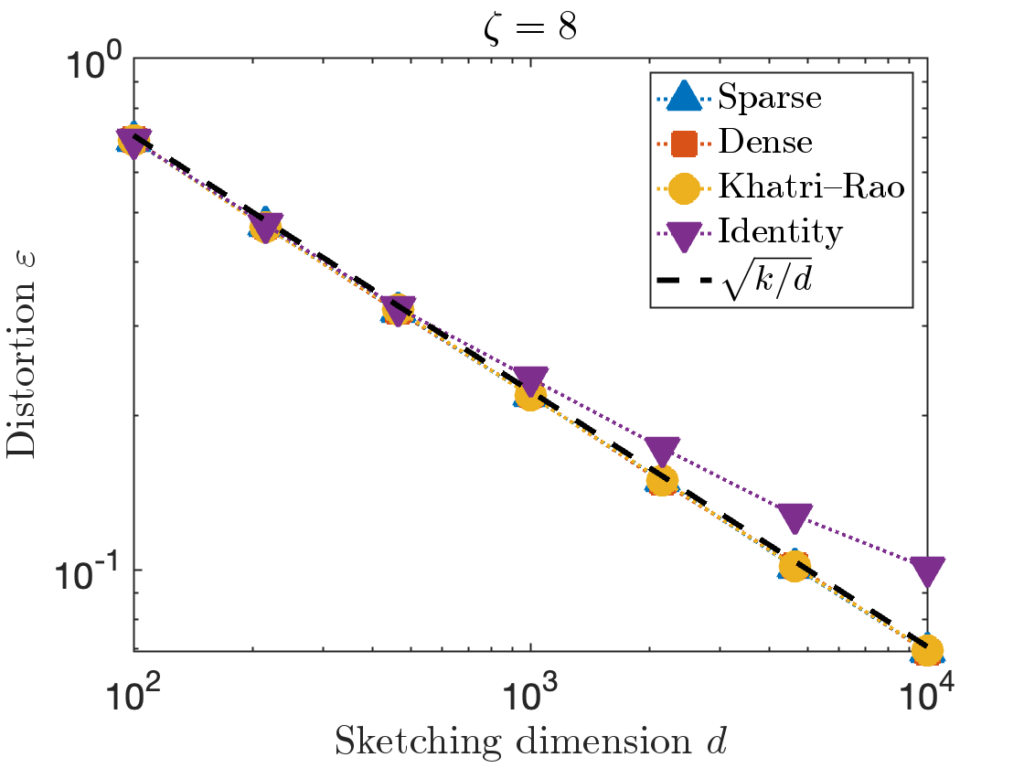
 . However, for the last test matrix “Identity”, we see the distortion begins to slightly exceed this predicted distortion for
. However, for the last test matrix “Identity”, we see the distortion begins to slightly exceed this predicted distortion for  .
. , we can increase the value of the sparsity parameter
, we can increase the value of the sparsity parameter  . We recommend
. We recommend ![\[\zeta = \max \left( 8 , \left\lceil 2\sqrt{\frac{d}{k}} \right\rceil \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5f1ebe7695d9918399b0f97b628fcce1_l3.png "Rendered by QuickLaTeX.com")
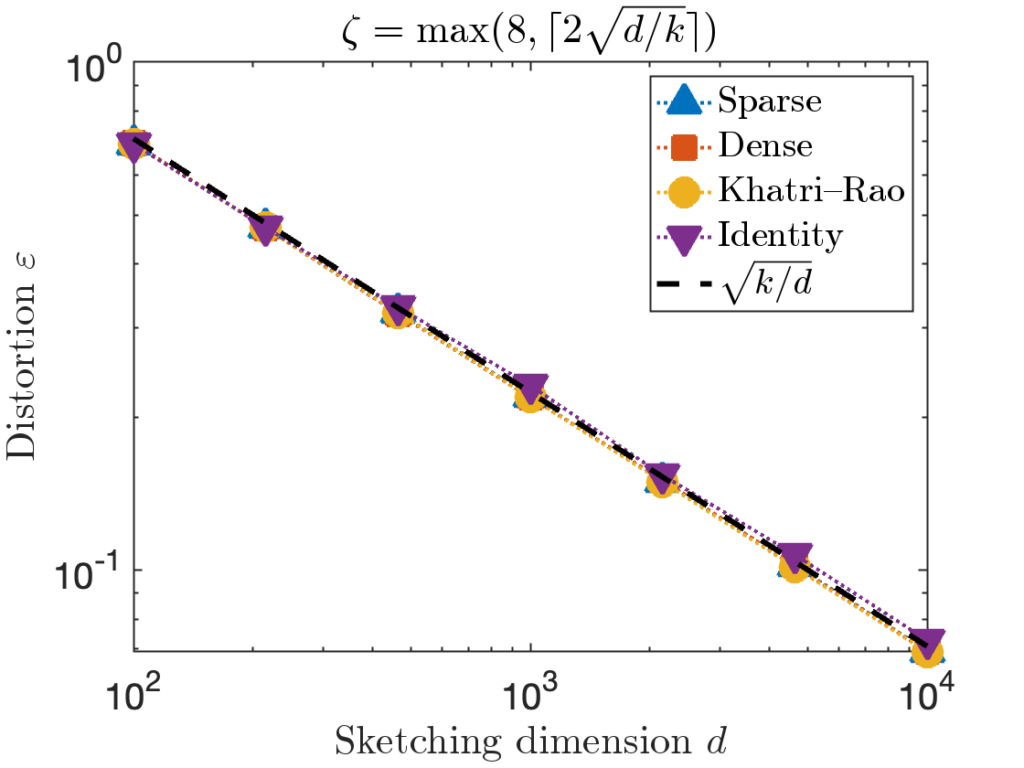
 can be necessary to achieve the optimal distortion.
can be necessary to achieve the optimal distortion.![\[S = \frac{1}{\sqrt{\zeta}} \begin{bmatrix} s_1 & \cdots & s_n \end{bmatrix}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4871e53b2bb59ad11946f2d1eafc66f7_l3.png "Rendered by QuickLaTeX.com")
 is an independent and randomly generated to contain exactly
is an independent and randomly generated to contain exactly ![\[(1-\varepsilon) \norm{x} \le \norm{Sx} \le (1+\varepsilon) \norm{x} \quad \text{for all }x \in \operatorname{col}(A).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-41fd14292c1c85cd318dde801a75e1bd_l3.png "Rendered by QuickLaTeX.com")
![\[d = \frac{k}{\varepsilon^2} \quad \text{and} \quad \zeta = \max\left(8,\frac{2}{\varepsilon}\right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ed85cec8dadffec7c7a3b4e099d6637d_l3.png "Rendered by QuickLaTeX.com")
 . The value
. The value  has demonstrated deficiencies and should almost always be avoided (see below). The scaling
has demonstrated deficiencies and should almost always be avoided (see below). The scaling  is derived from the
is derived from the ![\[d = \mathcal{O} \left( \frac{k \log k}{\varepsilon^2} \right) \quad \text{and} \quad \zeta = \mathcal{O}\left( \frac{\log k}{\varepsilon} \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4fb6d0d60c85bc8a60d95bb0a19e63d6_l3.png "Rendered by QuickLaTeX.com")
 factor and the lack of explicit constants in the
factor and the lack of explicit constants in the  and can also be generated using a single line. The real challenge to generating sparse sign embeddings in MATLAB is the row indices, since each batch of
and can also be generated using a single line. The real challenge to generating sparse sign embeddings in MATLAB is the row indices, since each batch of  sparse sign embedding with sparsity
sparse sign embedding with sparsity  and weights
and weights  . To apply
. To apply  and reweight them using the weights:
and reweight them using the weights:![\[b \in \real^n \longmapsto Sb = (w_1 b_{i_1},\ldots,w_db_{i_d}) \in \real^d.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-712b97fc6f6efb9bf9a40dac2a652f5c_l3.png "Rendered by QuickLaTeX.com")
 for the hard “Identity” test matrix used above.
for the hard “Identity” test matrix used above.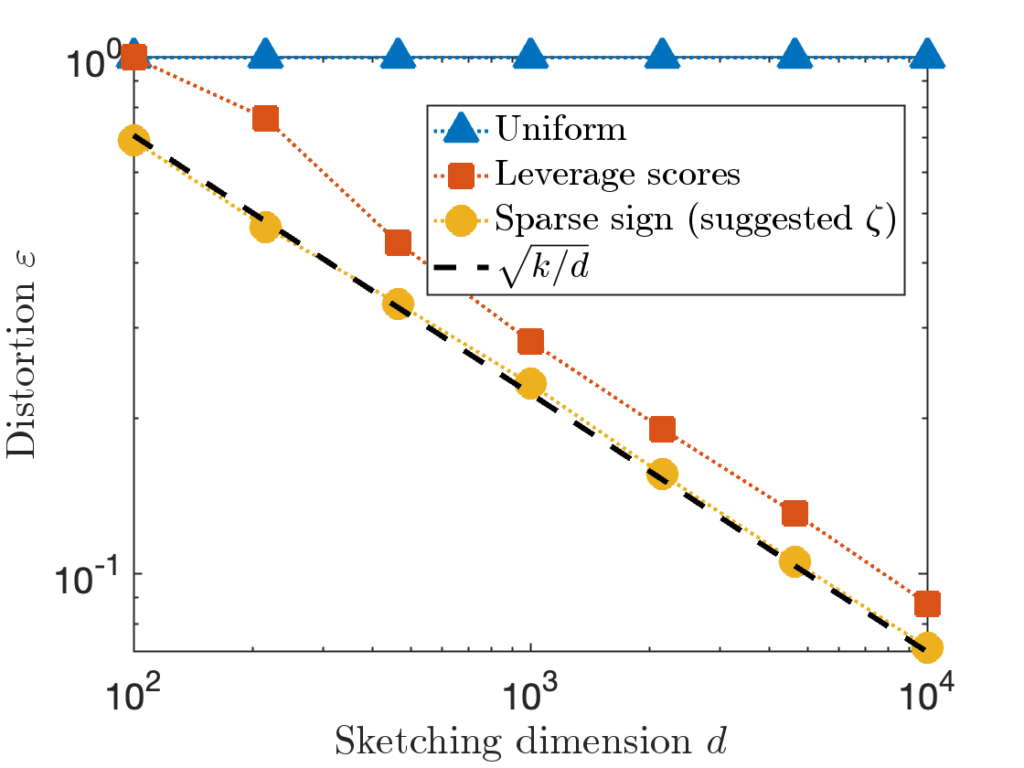
 for each row of
for each row of  cost, much higher than other types of sketches.
cost, much higher than other types of sketches. , and compare the distortion of CountSketch to the sparse sign embedding with parameters
, and compare the distortion of CountSketch to the sparse sign embedding with parameters  :
: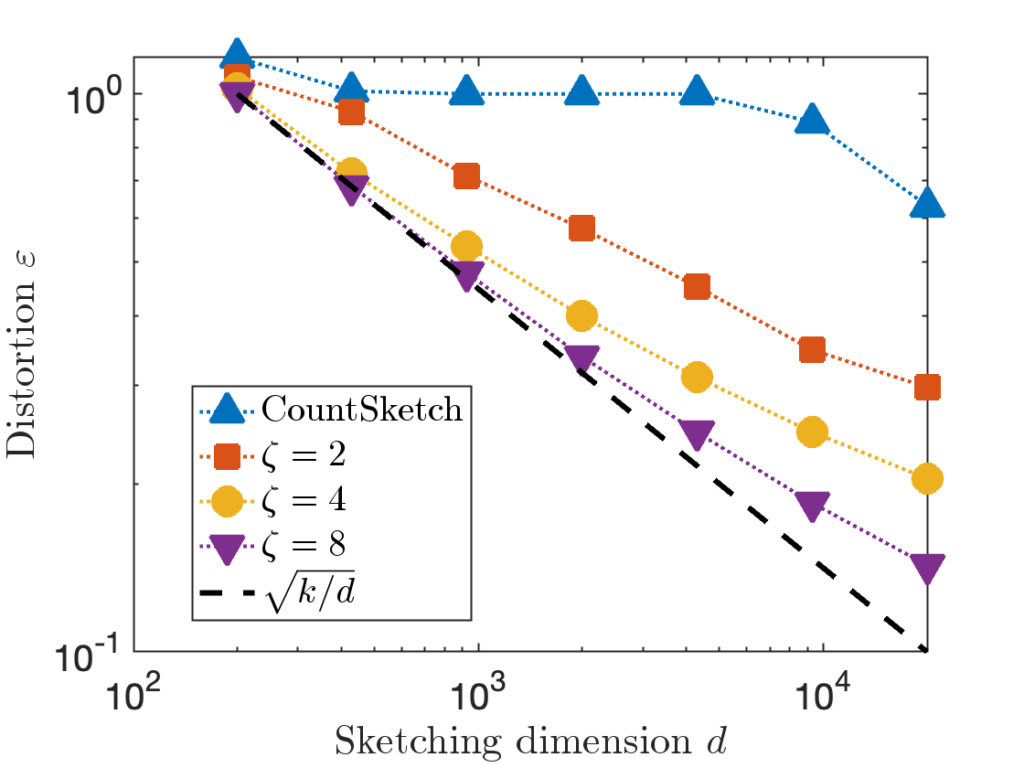
 , 20× higher than
, 20× higher than  in order to achieve distortion
in order to achieve distortion  (or perhaps
(or perhaps  ). This difference between
). This difference between  for CountSketch and
for CountSketch and  for other sketching matrices is a at the root of CountSketch’s woefully bad performance on some inputs.
for other sketching matrices is a at the root of CountSketch’s woefully bad performance on some inputs. is an informal symbol meaning “proportional to”.
is an informal symbol meaning “proportional to”. . This is Theorem 16 in
. This is Theorem 16 in  , where
, where  is a CountSketch of size
is a CountSketch of size  and
and  is a Gaussian sketching matrix of size
is a Gaussian sketching matrix of size  .
. is a CountSketch matrix with output dimension
is a CountSketch matrix with output dimension  , then the distortion of
, then the distortion of  with high probability.
with high probability.![\[SA = \begin{bmatrix} s_1 & \cdots & s_k \end{bmatrix}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-82c7ea0e5c52b6bd9bdb389f3d3f92d7_l3.png "Rendered by QuickLaTeX.com")
 has a single
has a single  in a uniformly random location
in a uniformly random location  .
.
 are not all different from each other, say
are not all different from each other, say  . Set
. Set  , where
, where  is the standard basis vector with
is the standard basis vector with  but
but  . Thus, for the distortion relation
. Thus, for the distortion relation ![\[(1-\varepsilon) \norm{x} =(1-\varepsilon)\sqrt{2} \le 0 = \norm{(SA)x}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b08feebb30829a0e73b02188eba6f901_l3.png "Rendered by QuickLaTeX.com")
 . Thus,
. Thus, ![\[\prob \{ \varepsilon \ge 1 \} \ge \prob \{ j_1,\ldots,j_k \text{ are not distinct} \}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-12fa7b716b97c7e9482f4f6327bd4728_l3.png "Rendered by QuickLaTeX.com")
 pairs of people. Each pair of people has a
pairs of people. Each pair of people has a  chance of sharing a birthday, so the expected number of birthdays in a room of 23 people is
chance of sharing a birthday, so the expected number of birthdays in a room of 23 people is  . Since are 0.69 birthdays shared on average in a room of 23 people, it is perhaps less surprising that 23 is the critical number at which the chance of two people sharing a birthday exceeds 50%.
. Since are 0.69 birthdays shared on average in a room of 23 people, it is perhaps less surprising that 23 is the critical number at which the chance of two people sharing a birthday exceeds 50%. and
and  in CountSketch have a
in CountSketch have a  chance of being the same. There are
chance of being the same. There are  pairs of indices, so the expected number of equal indices
pairs of indices, so the expected number of equal indices  . Thus, we should anticipate
. Thus, we should anticipate  is required to ensure that
is required to ensure that ![\[\prob \{ j_1,\ldots,j_k \text{ are not distinct} \} = 1 - \prob \{ j_1,\ldots,j_k \text{ are distinct} \}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-338a38501a565185b794f8ed03803c9a_l3.png "Rendered by QuickLaTeX.com")
 are all distinct, the probability
are all distinct, the probability  are distinct is just the probability that
are distinct is just the probability that  values
values ![\[\prob\{ j_1,\ldots,j_i \text{ are distinct} \mid j_1,\ldots,j_{i-1} \text{ are distinct}\} = 1 - \frac{i-1}{d}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b474f00c44e53b7c3c398d56121e1ac3_l3.png "Rendered by QuickLaTeX.com")
![\[\prob \{ j_1,\ldots,j_k \text{ are distinct} \} = \prod_{i=1}^k \left(1 - \frac{i-1}{d} \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7dd28281f8dbed8b3a1452a21e7410c8_l3.png "Rendered by QuickLaTeX.com")
 for every
for every  , obtaining
, obtaining![\[\mathbb{P} \{ j_1,\ldots,j_k \text{ are distinct} \} \le \prod_{i=0}^{k-1} \exp\left(-\frac{i}{d}\right) = \exp \left( -\frac{1}{d}\sum_{i=0}^{k-1} i \right) = \exp\left(-\frac{k(k-1)}{2d}\right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8d9d4c7d12af0fd89e53ca261c5df287_l3.png "Rendered by QuickLaTeX.com")
![\[\prob \{ \varepsilon \ge 1 \} \ge 1-\prob \{ j_1,\ldots,j_k \text{ are distinct} \\}\ge 1-\exp\left(-\frac{k(k-1)}{2d}\right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-144cfc73d778c4f9ed7442f5a6610e5e_l3.png "Rendered by QuickLaTeX.com")
![\[\prob\{\varepsilon \ge 1\} \ge \frac{1}{2} \quad \text{if} \quad d \le \frac{k(k-1)}{2\ln 2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e11567078a415b79b06a8e5e6f0b1600_l3.png "Rendered by QuickLaTeX.com")
 or perhaps a tall matrix
or perhaps a tall matrix  . A sketching matrix is a
. A sketching matrix is a  matrix
matrix  . When multiplied into a high-dimensional vector
. When multiplied into a high-dimensional vector 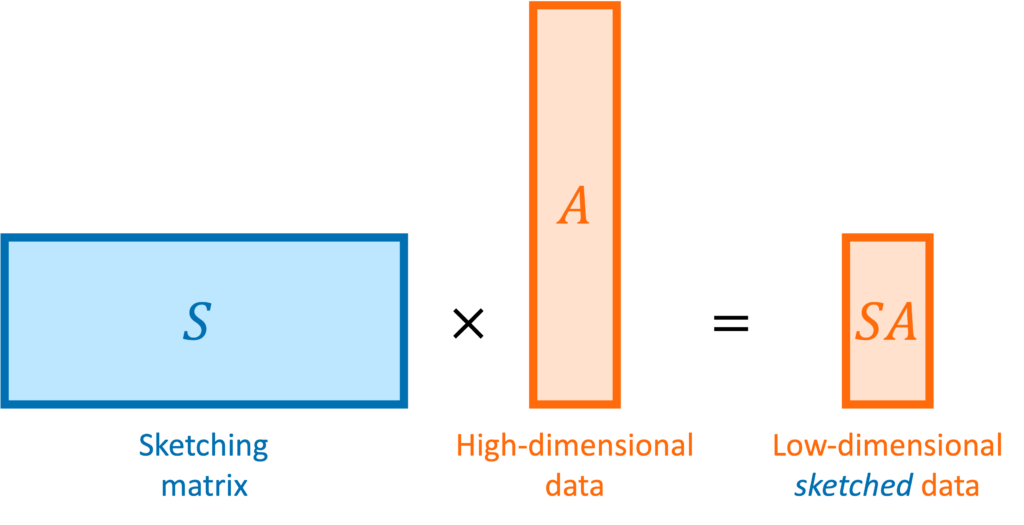
 be a collection of vectors. For
be a collection of vectors. For  , we require that
, we require that  :
: ![\[(1-\varepsilon) \norm{x}\le\norm{Sx}\le(1+\varepsilon)\norm{x} \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-735df49a45ff5d0bb27d478569004bdd_l3.png "Rendered by QuickLaTeX.com")
![\[\operatorname{col}(A) \coloneqq \{ Ax : x \in \real^k \}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5ecde6b43df29caba14fd43682b4cc86_l3.png "Rendered by QuickLaTeX.com")
 with an output dimension of roughly
with an output dimension of roughly  . In particular, the sketching dimension
. In particular, the sketching dimension  elements) or dimension (
elements) or dimension ( requires roughly
requires roughly  operations, rather than the
operations, rather than the  operations we would expect to multiply a
operations we would expect to multiply a  is
is ![\[S = \sqrt{\frac{n}{d}} \cdot R \cdot F \cdot D.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-92095cbf9c7d55273bd572a33debe415_l3.png "Rendered by QuickLaTeX.com")
 is a diagonal matrix whose entries are each a random
is a diagonal matrix whose entries are each a random  is a fast trigonometric transform such as a fast
is a fast trigonometric transform such as a fast  is a selection matrix. To generate
is a selection matrix. To generate  , let
, let  , selected without replacement.
, selected without replacement.  for every vector
for every vector  and the
and the  , and
, and  operations, a significant improvement over the
operations, a significant improvement over the  , larger than for a Gaussian sketch.
, larger than for a Gaussian sketch.![\[S = \frac{1}{\sqrt{\zeta}} \begin{bmatrix} s_1 & s_2 & \cdots & s_n \end{bmatrix}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-bb5806806e2e5900d4351c08befbd82e_l3.png "Rendered by QuickLaTeX.com")
 nonzero entries. The parameter
nonzero entries. The parameter  in practice.
in practice. or
or  operations) to apply to a vector, depending on parameter choices (see below). With a good sparse matrix library, sparse sign embeddings are often the fastest sketching matrix by a wide margin.
operations) to apply to a vector, depending on parameter choices (see below). With a good sparse matrix library, sparse sign embeddings are often the fastest sketching matrix by a wide margin. random numbers, higher than SRTTs (roughly
random numbers, higher than SRTTs (roughly  numbers).
numbers).  ; the theoretically sanctioned sketching dimension (at least according to existing theory) is larger than for a Gaussian sketch. In practice, we can often get away with using
; the theoretically sanctioned sketching dimension (at least according to existing theory) is larger than for a Gaussian sketch. In practice, we can often get away with using  .
. operations. Therefore, sketching offers the promise of speeding up linear algebraic computations involving
operations. Therefore, sketching offers the promise of speeding up linear algebraic computations involving ![\[\operatorname*{minimize}_{x\in\real^k} \norm{Ax - b}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-07528304ce875f54aa1173062c5ec10a_l3.png "Rendered by QuickLaTeX.com")
 . Applying
. Applying ![\[\operatorname*{minimize}_{\hat{x}\in\real^k} \norm{(SA)\hat{x} - Sb}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7110d3e9c1dd7e29793603a5bafdd201_l3.png "Rendered by QuickLaTeX.com")
 is the sketch-and-solve solution to the least-squares problem, which we can use as an approximate solution to the original least-squares problem.
is the sketch-and-solve solution to the least-squares problem, which we can use as an approximate solution to the original least-squares problem.
 , first apply sketching to obtain
, first apply sketching to obtain  and then apply an out-of-the-box clustering algorithms like
and then apply an out-of-the-box clustering algorithms like  denote the optimal least-squares solution and let
denote the optimal least-squares solution and let ![\[\norm{A\hat{x} - b} \le \frac{1+\varepsilon}{1-\varepsilon} \norm{Ax - b}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3d1d034bd8bf93f1c15e435a27a355ee_l3.png "Rendered by QuickLaTeX.com")
 , then this bound tells us that
, then this bound tells us that ![\[\norm{A\hat{x} - b} \le 2\norm{Ax_\star - b}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-588170743415f491f48ef2adc3d46628_l3.png "Rendered by QuickLaTeX.com")
 . For such applications, the bound (4) ensures that
. For such applications, the bound (4) ensures that  . Often, this means
. Often, this means  , measuring how close
, measuring how close  and residual norm
and residual norm  ). Then, we generate a sparse sign embedding of dimension
). Then, we generate a sparse sign embedding of dimension  ). Then, we compute the sketch-and-solve solution and, as reference, a “direct” solution by
). Then, we compute the sketch-and-solve solution and, as reference, a “direct” solution by  , close to direct method’s residual norm of
, close to direct method’s residual norm of  . However, the forward error of sketch-and-solve is
. However, the forward error of sketch-and-solve is  nine orders of magnitude larger than the direct method’s forward error of
nine orders of magnitude larger than the direct method’s forward error of  .
. , to decrease the distortion by a factor of ten requires increasing the sketching dimension
, to decrease the distortion by a factor of ten requires increasing the sketching dimension  that we hope will converge at a rapid rate to the true least-squares solution,
that we hope will converge at a rapid rate to the true least-squares solution,  , then
, then  .
.![\[SA = QR,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9edf84077821e9e0cd0c772b77024799_l3.png "Rendered by QuickLaTeX.com")
![\[A^\top A \approx (SA)^\top (SA) = R^\top Q^\top Q R = R^\top R.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-90e14ee1172525fd795980cb3ef1a647_l3.png "Rendered by QuickLaTeX.com")
 since
since  has orthonormal columns. The conclusion is that
has orthonormal columns. The conclusion is that  .
.
![\[(A^\top A)x = A^\top b. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3f3c5265345a169d6633a759854510da_l3.png "Rendered by QuickLaTeX.com")
 by
by  in (5) and solving. The resulting solution is
in (5) and solving. The resulting solution is![\[x_0 = R^{-1} (R^{-\top}(A^\top b)).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6b63093ea5c1d9714fb0deab43fc4dc3_l3.png "Rendered by QuickLaTeX.com")
 will typically not be a good solution to the least-squares problem (2), so we need to iterate. To do so, we’ll try and solve for the error
will typically not be a good solution to the least-squares problem (2), so we need to iterate. To do so, we’ll try and solve for the error  . To derive an equation for the error, subtract
. To derive an equation for the error, subtract  from both sides of the normal equations (5), yielding
from both sides of the normal equations (5), yielding ![\[(A^\top A)(x-x_0) = A^\top (b-Ax_0).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4f1ee993844dbcd8e397f27ccd1deefb_l3.png "Rendered by QuickLaTeX.com")
 :
: ![\[x\approx x_1 \coloneqq x_0 + R^{-\top}(R^{-1}(A^\top(b-Ax_0))).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0496d7e3c8bc117e658c57fc167081be_l3.png "Rendered by QuickLaTeX.com")
 , approximate
, approximate  , and obtain a new approximate solution
, and obtain a new approximate solution  . Continuing this process, we obtain an iteration
. Continuing this process, we obtain an iteration![\[x_{i+1} = x_i + R^{-\top}(R^{-1}(A^\top(b-Ax_i))).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f8db238fc978e6a8fc968fc5e387ec27_l3.png "Rendered by QuickLaTeX.com")
 at every iteration. Later,
at every iteration. Later, 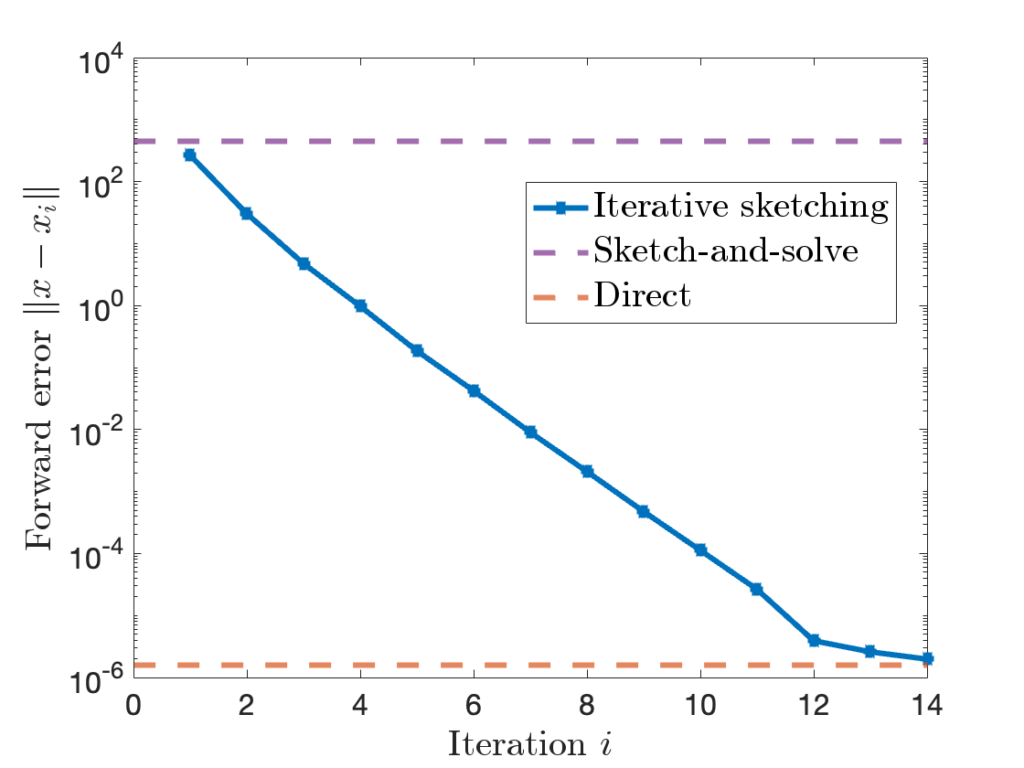
 to
to  , the accuracy of the iterative sketching solution
, the accuracy of the iterative sketching solution  can be quite high.
can be quite high.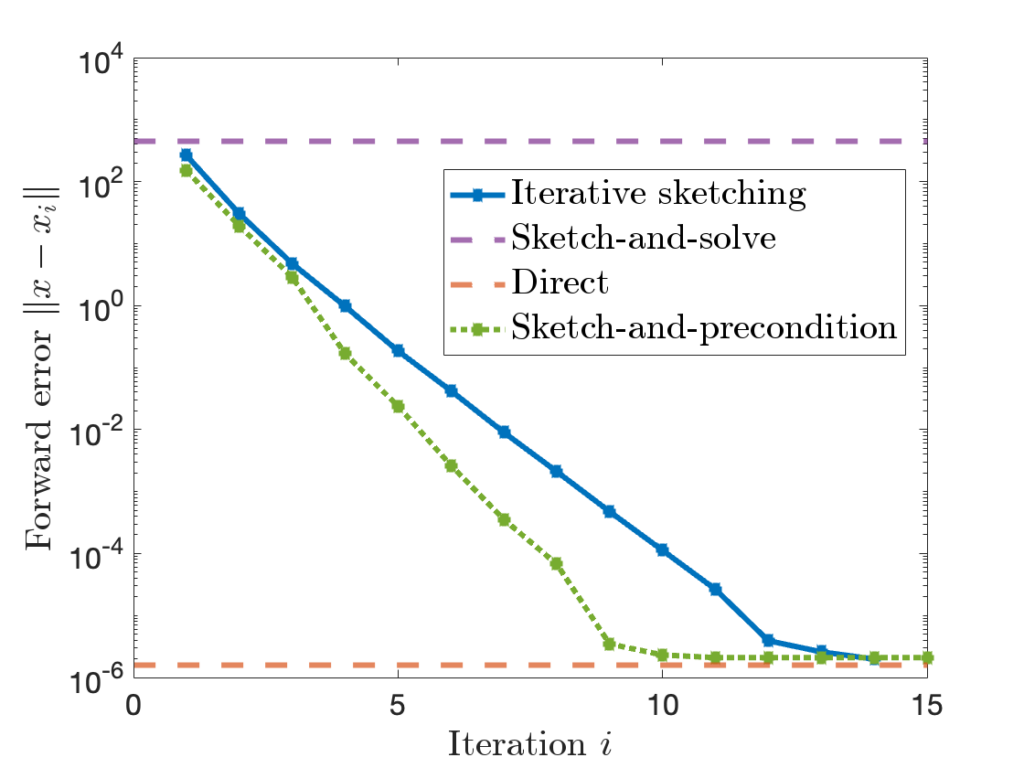
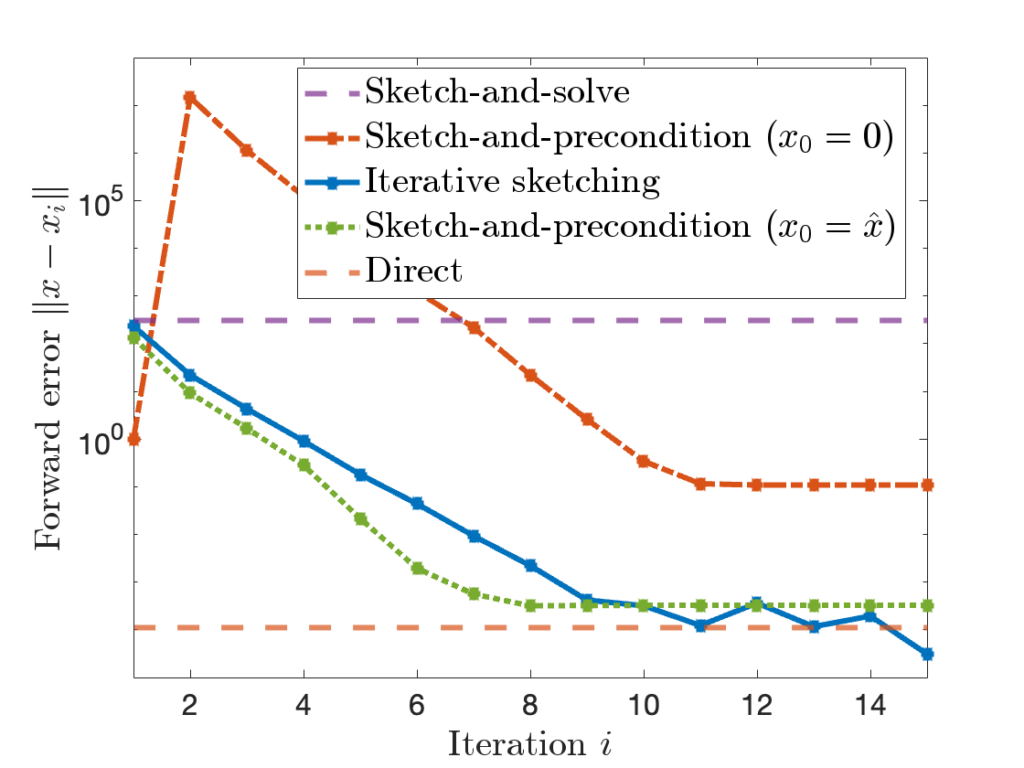
 is comparable to accuracy of a standard
is comparable to accuracy of a standard  , sketch-and-precondition is not forward stable. The maximum achievable accuracy was worse than standard solvers by orders of magnitude! Maybe sketching doesn’t work after all?
, sketch-and-precondition is not forward stable. The maximum achievable accuracy was worse than standard solvers by orders of magnitude! Maybe sketching doesn’t work after all? for sketch-and-precondition, then sketch-and-precondition appears to be forward stable in practice. No theoretical analysis supporting this finding is known at present.
for sketch-and-precondition, then sketch-and-precondition appears to be forward stable in practice. No theoretical analysis supporting this finding is known at present. and residual
and residual  .
.
![\[\chi^2\left(\rho^{(n)} \, \middle|\middle| \, \pi\right) \le \left( \max \{ \lambda_2, -\lambda_n \} \right)^{2n} \chi^2\left(\rho^{(0)} \, \middle|\middle| \, \pi\right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ed65d0043f7a436b0f42a6d096d34e9f_l3.png "Rendered by QuickLaTeX.com")
 denotes the distribution of the chain at time
denotes the distribution of the chain at time  denotes the
denotes the  denotes the
denotes the 
 denote the decreasingly ordered
denote the decreasingly ordered  . To bound the rate of convergence to stationarity, we therefore must upper bound
. To bound the rate of convergence to stationarity, we therefore must upper bound  and lower bound
and lower bound  .
.
 Fortunately, there is a trick.
Fortunately, there is a trick. for every
for every  , where
, where  denotes the identity matrix. It is easy to see that the stationary distribution
denotes the identity matrix. It is easy to see that the stationary distribution  :
:![\[\pi^\top P^{\rm lazy} = \frac{1}{2} \pi^\top P + \frac{1}{2} \pi^\top I = \frac{1}{2} \pi^\top + \frac{1}{2} \pi^\top = \pi^\top.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d33d9a3024c655134a56d89b119cf39c_l3.png "Rendered by QuickLaTeX.com")
![\[\lambda_i^{\rm lazy} = \frac{1+\lambda_i}{2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ac2938aa6854a708a394222649af1e66_l3.png "Rendered by QuickLaTeX.com")
 are
are  , all of the eigenvalues of the lazy chain are
, all of the eigenvalues of the lazy chain are  . Thus, the smallest eigenvalue of
. Thus, the smallest eigenvalue of  :
:![\[\chi^2\left(\rho^{(n)} \, \middle|\middle| \, \pi\right) \le \left( \lambda_2^{\rm lazy} \right)^{2n} \chi^2\left(\rho^{(0)} \, \middle|\middle| \, \pi\right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-861a132f08a1c2b208d61eb004ed58bb_l3.png "Rendered by QuickLaTeX.com")
 over a random variable
over a random variable  drawn from the stationary distribution:
drawn from the stationary distribution: ![\[\expect_{x\sim \pi} [f(x)] = \sum_{i=1}^m f(i) \pi_i.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6399c58f0e1b13f3cffac439c145d3b4_l3.png "Rendered by QuickLaTeX.com")
 of the value of
of the value of  values
values  of the chain
of the chain![\[\hat{f}_{N}\coloneqq \frac{1}{N}\sum_{n=0}^{N-1} f(x_i).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-591e83a4f5551f6826ddffb5de03ce72_l3.png "Rendered by QuickLaTeX.com")
![\[\hat{f}_N = \frac{1}{N}\sum_{n=0}^{N-1} f(x_i)\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-615239005583b665db04e6a578abec08_l3.png "Rendered by QuickLaTeX.com")
 denote the states of a Markov chain initialized in the stationary distribution
denote the states of a Markov chain initialized in the stationary distribution  . For a large number
. For a large number ![\expect_{x\sim \pi} [f(x)]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e4b2ff50078922c10c1f0a32237a19d6_l3.png "Rendered by QuickLaTeX.com") and variance
and variance  where
where ![\[\sigma^2 = \Var[f(x_0)] + 2\sum_{n=1}^\infty \Cov(f(x_0),f(x_n)). \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b209aff956ed442df57ac781c0376a2f_l3.png "Rendered by QuickLaTeX.com")
 and later values
and later values  . The faster the covariance decreases, the smaller
. The faster the covariance decreases, the smaller  will be and thus the smaller the error for the Markov chain average.
will be and thus the smaller the error for the Markov chain average. ,
, ![\[\frac{\hat{f}_N - \expect_{x \sim \pi} [f(x)]}{\sqrt{N}}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f5818a8980215e63da79048168aa8b79_l3.png "Rendered by QuickLaTeX.com")
 be any function.
be any function. that are
that are ![\[\langle \varphi_i,\varphi_j \rangle = \expect_{x\sim \pi} [\varphi_i(x)\varphi_j(x)] = \begin{cases}1, & i = j, \\0, & i \ne j.\end{cases}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-139934705e34440f3c903f0e089f580b_l3.png "Rendered by QuickLaTeX.com")
 as defining a function
as defining a function  . Thus, we can expand the function
. Thus, we can expand the function ![\[f = c_1 \varphi_1 + \cdots + c_m \varphi_m. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c7d7e27ce6d2d6d0e3c721a120eef469_l3.png "Rendered by QuickLaTeX.com")
 is the vector of all ones (or, equivalently, the function that outputs
is the vector of all ones (or, equivalently, the function that outputs  at time
at time  at time
at time  ?
? conditional on the chain starting at
conditional on the chain starting at  :
:![\[(Pf)(i) = \expect[f(x_1) \mid x_0 = i].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d3abc35eacbcac194f948451539f7778_l3.png "Rendered by QuickLaTeX.com")
![\[(P^nf)(i) = \expect[f(x_n) \mid x_0 = i]. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3aee313626d01d020c481e25567fe6c1_l3.png "Rendered by QuickLaTeX.com")
 denote a probability distribution which places 100% of the probability mass on the single site
denote a probability distribution which places 100% of the probability mass on the single site  is
is ![\[(P^n f)(i) = \delta_i^\top (P^n f) = (\delta_i^\top P^n) f.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e8c9b07d76e51bf1a6c2e582e9a239a7_l3.png "Rendered by QuickLaTeX.com")
 is the state of the Markov chain after
is the state of the Markov chain after  . Thus,
. Thus,![\[(P^n f)(i) = (\rho^{(n)})^\top f = \sum_{i=1}^m \rho^{(n)}_i f(i) = \expect[f(x_n) \mid x_0=i].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6eaa69991373f0004f94c020fd9d4b3c_l3.png "Rendered by QuickLaTeX.com")
 and the spectral decomposition of
and the spectral decomposition of  . Then
. Then![\[\Cov (f(x_0),f(x_n)) = \expect[f(x_0)f(x_n)] - \expect[f(x_0)]\expect[f(x_n)]. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0d2881b12745eed9e1a985eb8fefa650_l3.png "Rendered by QuickLaTeX.com")
![\expect[f(x_0)]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c88debcfbbae714aadb6b34e5a6fd646_l3.png "Rendered by QuickLaTeX.com") and
and ![\expect[f(x_n)]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-35ed314f41c6654ee1ef7f9631c96e4e_l3.png "Rendered by QuickLaTeX.com") . Since
. Since ![\[\expect[f(x_0)] = \expect[f(x_0) \cdot 1] = \expect[f(x_0) \varphi_1(x_0)] = \langle f, \varphi_1\rangle = c_1.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-fceebd56048f584bab0bf0ba12254652_l3.png "Rendered by QuickLaTeX.com")
 .
.
 , so we have
, so we have ![\expect[f(x_n)] = \expect[f(x_0)] = c_1](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-703a86f4cdb9f7d354b2f7440826b5e9_l3.png "Rendered by QuickLaTeX.com") .
.![\expect[f(x_0)f(x_n)]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-274aba259d8ec1c94a05588a5b6e3846_l3.png "Rendered by QuickLaTeX.com") . Use the
. Use the ![\begin{align*}\expect[f(x_0)f(x_n)] &= \sum_{i=1}^m \expect[f(x_0) f(x_n) \mid x_0 = i] \prob\{x_0 = i\} \\&= \sum_{i=1}^m f(i) \expect[f(x_n) \mid x_0 = i] \pi_i.\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2e350d683f2a7cfff71028de3a45dbf8_l3.png "Rendered by QuickLaTeX.com")
![\[\expect[f(x_0)f(x_n)] = \sum_{i=1}^m f(i) (P^n f)(i) \pi_i = \langle f, P^n f\rangle.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-10a769b03dee2b18b03876d46eaac7c7_l3.png "Rendered by QuickLaTeX.com")
![\[\expect[f(x_0)f(x_n)] = \left\langle \sum_{i=1}^m c_i \varphi_i,\sum_{i=1}^m c_i P^n\varphi_i \right\rangle = \left\langle \sum_{i=1}^m c_i \varphi_i,\sum_{i=1}^m c_i \lambda_i^n \varphi_i \right\rangle = \sum_{i=1}^m \lambda_i^n \, c_i^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-63bbc0c0f98d4c57de9a53a7162295f3_l3.png "Rendered by QuickLaTeX.com")
![\expect[f(x_0)] = \expect[f(x_n)] = c_1](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0dee583b20d0c65301633cb289069661_l3.png "Rendered by QuickLaTeX.com") and plugging into (4), we obtain
and plugging into (4), we obtain![\[\Cov(f(x_0),f(x_n)) = \sum_{i=1}^m \lambda_i^n \, c_i^2 - c_1^2 = \sum_{i=2}^m \lambda_i^n c_i^2. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-20b7af4f300077ae2d041dae30de529d_l3.png "Rendered by QuickLaTeX.com")
 , so
, so  entirely drops out of the covariance.
entirely drops out of the covariance.
![\[\Var[f(x_0)] = \Cov(f(x_0),f(x_0)) = \sum_{i=2}^m \lambda_i^0 c_i^2 = \sum_{i=2}^m c_i^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5f587d4d05f65d8d43d5824fd499d0d2_l3.png "Rendered by QuickLaTeX.com")
![\begin{align*}\sigma^2 &= \Var[f(x_0)] + 2\sum_{n=1}^\infty \Cov(f(x_0),f(x_n)) \\&= \sum_{i=2}^m c_i^2 + 2\sum_{n=1}^\infty \sum_{i=2}^m \lambda_i^n \, c_i^2 \\&= -\sum_{i=2}^m c_i^2 + 2\sum_{i=2}^m \left(\sum_{n=0}^\infty \lambda_i^n\right)c_i^2 .\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-da05968b01086497f8e6e098e384b170_l3.png "Rendered by QuickLaTeX.com")
![\[\sigma^2 = -\sum_{i=2}^m c_i^2 + 2\sum_{i=2}^m \frac{1}{1-\lambda_i} c_i^2 = \sum_{i=2}^m \left(\frac{2}{1-\lambda_i}-1\right)c_i^2 = \sum_{i=2}^m \frac{1+\lambda_i}{1-\lambda_i} c_i^2. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-61c9cb2ae48b8b474edd697e36a1e58c_l3.png "Rendered by QuickLaTeX.com")
![\[\hat{f}_N = \frac{1}{N} \sum_{i=0}^{N-1} f(x_n)\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-76330a2c8101f63831cdc0bcdd4e0cee_l3.png "Rendered by QuickLaTeX.com")
![\expect_{x \sim \pi} [f(x)]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c4b525a230a1b1912a79546d8b641e00_l3.png "Rendered by QuickLaTeX.com") . The Markov chain central limit theorem shows that, for a large number of steps
. The Markov chain central limit theorem shows that, for a large number of steps ![\hat{f}_N - \expect_{x\sim\pi}[f(x)]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0f260b35378f3e3cb9ca1723ad1a0fe1_l3.png "Rendered by QuickLaTeX.com") is approximately normally distributioned with mean zero and variance
is approximately normally distributioned with mean zero and variance  of the transition matrix
of the transition matrix  . Thus, we have
. Thus, we have
 of an eigenvalue
of an eigenvalue  is scaled by in
is scaled by in  for the corresponding eigenvalue
for the corresponding eigenvalue  of the lazy chain. We see that at every
of the lazy chain. We see that at every  value, the lazy chain has a higher amplification factor than the original chain.
value, the lazy chain has a higher amplification factor than the original chain.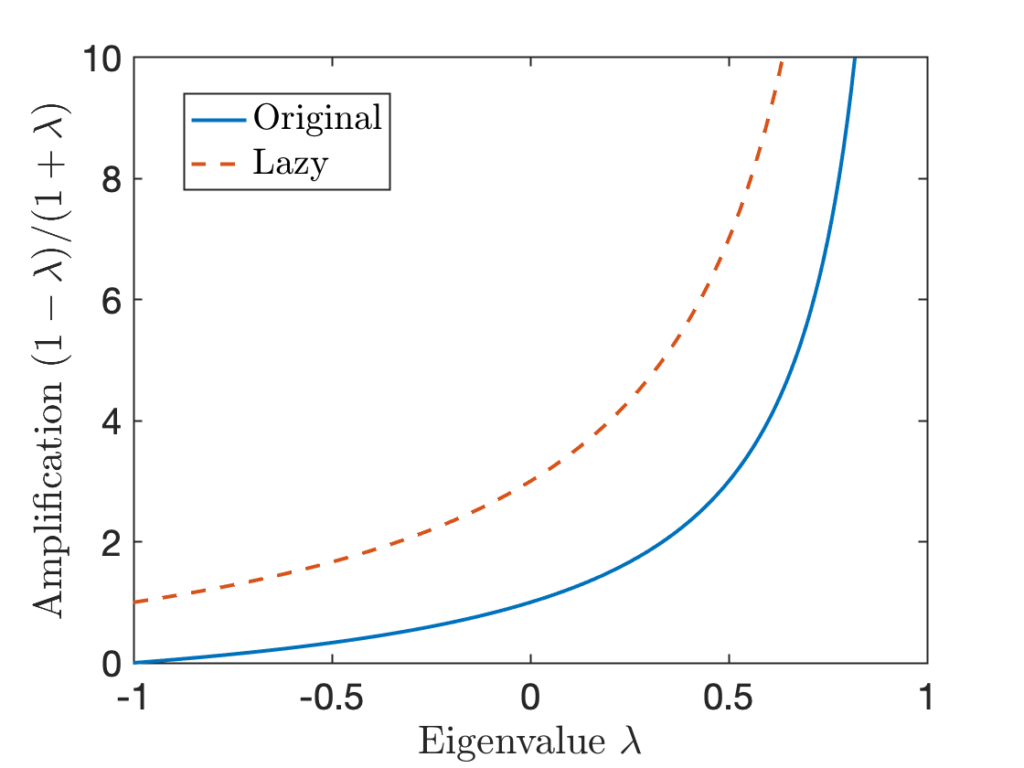
 with transition probability matrix
with transition probability matrix  and
and  and functions
and functions  as being one and the same, and we will use both
as being one and the same, and we will use both  and
and  to denote the
to denote the  with a numeric value.
with a numeric value.![\expect[f]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-fb89f41552bdb76a7c8c04d9d35b3631_l3.png "Rendered by QuickLaTeX.com") denote the
denote the  where
where ![\[\expect[f] \coloneqq \expect_{X \sim \pi} [f(X)] = \sum_{i=1}^m \pi_i f(i).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-09e6ff4df06017b52b788b6787fad904_l3.png "Rendered by QuickLaTeX.com")
![\[\Var(f) \coloneqq \expect [(f - \expect[f])^2] = \sum_{i=1}^m (f(i) - \expect[f])^2 \pi_i.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-03395d1f27b782837abde9a7d030c4c7_l3.png "Rendered by QuickLaTeX.com")
 denote a probability distribution which assigns 100% probability to outcome
denote a probability distribution which assigns 100% probability to outcome  matrix with real entries
matrix with real entries  ) has
) has  .
. . Let’s first ask: What does the transpose
. Let’s first ask: What does the transpose  is equipped with the
is equipped with the  and it is defined as
and it is defined as![\[(f,g) \coloneqq f^\top g = \sum_{i=1}^m f(i) g(i).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-86465508e63dc4f1aefde253dd2b2a7c_l3.png "Rendered by QuickLaTeX.com")
![\[(f,Ag) = (A^\top f, g)\quad \text{for every }f,g\in\real^m.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9bc76899fbef0450c0757d9d90e3722a_l3.png "Rendered by QuickLaTeX.com")
 is the same as the amount of
is the same as the amount of  in the direction of
in the direction of ![[\cdot,\cdot]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9f5a656d3836fd9f84831aee0283adc1_l3.png "Rendered by QuickLaTeX.com") be any inner product on
be any inner product on  such that
such that ![\[[f,Ag]=[A^*f,g]\quad\text{for every }f,g\in\real^m.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-fe7c2788bf80274e17adc7bef25f9701_l3.png "Rendered by QuickLaTeX.com")
 .
.![\[[u_i,u_j]=\begin{cases}1, & i=j, \\0,& i\ne j.\end{cases}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6485908bc8e69e0ad6fb16b826344f60_l3.png "Rendered by QuickLaTeX.com")
![\[\langle f, g \rangle \coloneqq \expect[f\cdot g] = \sum_{i=1}^m f(i)g(i) \pi_i.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a1d97adbf15c3c5bd85460e94161ee28_l3.png "Rendered by QuickLaTeX.com")
![\expect[f] = \expect[g] = 0](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-16f996fb8f734dee3bcc58dc1b6638b1_l3.png "Rendered by QuickLaTeX.com") ), it reports the
), it reports the  where
where  is drawn from
is drawn from  :
:
![\[\pi_i P_{ij} = \pi_j P_{ji} \quad \text{for } i,j=1,2,\ldots,m\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d24fd7216a2b9a2afc49c2f1ae9ea55c_l3.png "Rendered by QuickLaTeX.com")
![\[\langle f, Pg \rangle= \sum_{i,j=1}^m f(i) g(j) \pi_jP_{ji} = \sum_{j=1}^m \left( \sum_{i=1}^m f(i) P_{ij} \right) g(j) \pi_j = \langle Pf, g\rangle.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2e8d0788b8c0f3ee0e4602e817957cfa_l3.png "Rendered by QuickLaTeX.com")
 associated with eigenvectors
associated with eigenvectors  which are orthonormal in the
which are orthonormal in the ![\[\langle\varphi_i,\varphi_j\rangle=\begin{cases}1, &i=j\\0,& i\ne j.\end{cases}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-fd0e99bc2439329beccce1339db0aee9_l3.png "Rendered by QuickLaTeX.com")
 span all of
span all of  elsewhere. Then, by the fundamental theorem of Markov chains,
elsewhere. Then, by the fundamental theorem of Markov chains,  converges to
converges to  for every
for every  ,
,  . Thus, since
. Thus, since  for every vector
for every vector  in magnitude.
in magnitude. is a vector of all one’s.
is a vector of all one’s. have magnitude
have magnitude  .
. for every
for every  .
. and
and ![\[\norm{\sigma - \pi}_{\rm TV} = \max_{A \subseteq \{1,\ldots,m\}} |\sigma(A) - \pi(A)| = \frac{1}{2} \sum_{i=1}^m |\sigma_i - \pi_i|.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d369d587213cfe7bb579ea5d2d89d1f8_l3.png "Rendered by QuickLaTeX.com")

 and
and  of each outcome. Spectral theory plays more nicely with an “
of each outcome. Spectral theory plays more nicely with an “ ” way of comparing probability distributions, which we develop now.
” way of comparing probability distributions, which we develop now. . This provides a general way of figuring out which objects for finite state space Markov chains are row vectors and which are column vectors: Measures are row vectors whereas functions are column vectors.
. This provides a general way of figuring out which objects for finite state space Markov chains are row vectors and which are column vectors: Measures are row vectors whereas functions are column vectors. given by
given by![\[h(i) = \frac{d\sigma}{d\pi}(i) = \frac{\sigma_i}{\pi_i} \quad \text{for } i=1,2,\ldots,m.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-cbffacfb7f8993bd3f61752e12035912_l3.png "Rendered by QuickLaTeX.com")
![\[\expect_{X \sim \sigma} [g(X)] = \expect_{Y \sim \pi}[g(Y)h(Y)] = \sum_{i=1}^m g(i)h(i) \pi_i. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b0bf11be5c7b4508cf0965a0c53426ab_l3.png "Rendered by QuickLaTeX.com")
 is a random variable with distribution
is a random variable with distribution ![\mathrm{Unif}[0,1]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0db92c7166ac7b273b85e4c488d226ea_l3.png "Rendered by QuickLaTeX.com") ) is a function
) is a function ![\[\expect_{X \sim \sigma} [g(X)] = \expect_{Y\sim \mathrm{Unif}[0,1]} [g(Y)h(Y)] = \int_0^1 g(x) h(x) \, dx.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c7f33161d65ad1fcb0f79f24b4f51a0f_l3.png "Rendered by QuickLaTeX.com")
 replaced with integrals
replaced with integrals  .
.
![\[\chi^2(\sigma \mid\mid \pi) \coloneqq \Var \left(\frac{d\sigma}{d\pi} \right) = \expect \left[\left( \frac{d\sigma}{d\pi} - 1 \right)^2\right] = \expect \left[\left(\frac{d\sigma}{d\pi}\right)^2\right] - 1.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2521b63ae50f02fe6099c54dcc4e00a4_l3.png "Rendered by QuickLaTeX.com")
 , note that
, note that![\[\expect\left[\frac{d\sigma}{d\pi}\right] = \sum_{i=1}^m \frac{d\sigma}{d\pi}(i) \pi_i = \sum_{i=1}^m \frac{\sigma_i}{\pi_i} \pi_i = \sum_{i=1}^m \sigma_i = 1. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6bcec4a11dcc43c0b01738d12e38d6e6_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{\sigma - \pi}_{\rm TV} = \frac{1}{2} \expect \left[ \left| \frac{d\sigma}{d\pi} - 1 \right| \right].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4ab771f36e4aabcf1fcbe807729630b6_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{\sigma - \pi}_{\rm TV} = \frac{1}{2} \expect \left[ \left| \frac{d\sigma}{d\pi} - 1 \right| \right] \le \frac{1}{2} \left(\expect \left[ \left( \frac{d\sigma}{d\pi} - 1 \right)^2 \right]\right)^{1/2} = \frac{1}{2} \sqrt{\chi^2(\sigma \mid\mid \pi)}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4986b7434b84b79aff6f94df2ec4b4e8_l3.png "Rendered by QuickLaTeX.com")
 denote the distributions of the Markov chain at times
denote the distributions of the Markov chain at times  . Then
. Then and
and  ,
, ![\[\chi^2(\delta_i \mid\mid \pi) \le \frac{1}{\pi_i}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4896a0499c8c3b1ab5bc1e2952fd1d65_l3.png "Rendered by QuickLaTeX.com")
![\[\chi^2\left( \rho^{(0)} \, \middle|\middle| \, \pi \right) \le \frac{1}{\min_{1\le i\le m} \pi_i} \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c3a7cb7312400b0230e1a53951e6d0a7_l3.png "Rendered by QuickLaTeX.com")
![\[\chi^2(\delta_i \mid\mid \pi) = \Var(d\delta_i/d\pi) \le \expect[(d\delta_i/d\pi)^2] = 1/\pi_i.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7343459c314bf5425643e99e6f0e2906_l3.png "Rendered by QuickLaTeX.com")
 is a
is a  for some
for some ![\[\norm{\rho^{(n)} - \pi}_{\rm TV} \le \varepsilon\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5ed1f6d26bba1fa54f9bd01bcf39defc_l3.png "Rendered by QuickLaTeX.com")
![\[n = \left\lceil \frac{1}{\log(\max\{\lambda_2,-\lambda_n\})}\log \left( \frac{1}{2\varepsilon \min_{1\le i \le m}\sqrt{\pi_i}} \right) \right\rceil \text{ steps}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8d9dc3295020a8171795dec7e448eef5_l3.png "Rendered by QuickLaTeX.com")

 . To do this, we use the recurrence for the probability distribution:
. To do this, we use the recurrence for the probability distribution:![\[\rho^{(n+1)}_j = \sum_{i=1}^m \rho_i^{(n)} P_{ij}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-84dcc77d0a4138d7d59160bcf259be32_l3.png "Rendered by QuickLaTeX.com")
![\[\left( \frac{d\rho^{(n+1)}}{d\pi}\right)_j = \frac{\rho_j^{(n+1)}}{\pi_j} = \frac{\sum_{i=1}^m \rho^{(n)}_i P_{ij}}{\pi_j}= \sum_{i=1}^m \rho^{(n)}_i\frac{P_{ij}}{\pi_j}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b6c60937b23d2509e4a3e4fa1f121427_l3.png "Rendered by QuickLaTeX.com")
 , which implies
, which implies![\[\left( \frac{d\rho^{(n+1)}}{d\pi}\right)_j = \sum_{i=1}^m\rho^{(n)}_i \frac{P_{ji}}{\pi_i} = \sum_{i=1}^m P_{ji} \frac{\rho^{(n)}_i}{\pi_i} = \left( P \frac{d \rho^{(n)}}{d\pi} \right)_j.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d22315b80e3fc4d3736d0c49f877fa9c_l3.png "Rendered by QuickLaTeX.com")
 is an ordinary
is an ordinary ![\[\frac{d\rho^{(n+1)}}{d\pi} = P \frac{d\rho^{(n)}}{d\pi} \quad \text{for } n =0,1,\ldots. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-406471ab024dae5551eae8b499a0243e_l3.png "Rendered by QuickLaTeX.com")
![\[\frac{d\rho^{(0)}}{d\pi} = c_1 \mathbf{1} + c_2 \varphi_2 + \cdots + c_m \varphi_m.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6bcb8bbdd130a3e85c422adc2f37715d_l3.png "Rendered by QuickLaTeX.com")
![\expect[d\rho^{(0)}/d\pi] = 1](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-37f5774410e7d60045c0758953e0cdde_l3.png "Rendered by QuickLaTeX.com") . Thus, we conclude
. Thus, we conclude  . Since the
. Since the  are eigenvectors of
are eigenvectors of  , the recurrence (7) implies
, the recurrence (7) implies![\[\frac{d\rho^{(n)}}{d\pi} = \mathbf{1} + c_2 \lambda_2^n \varphi_2 + \cdots + c_m \lambda_m^n \varphi_m.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-935cf156fcf1f467b0c6719614b94e6b_l3.png "Rendered by QuickLaTeX.com")
![\[\chi^2(\rho^{(n)} \mid\mid \pi) = \sum_{i=2}^m c_i^2 \lambda_i^{2n} \le (\max \{ \lambda_2, -\lambda_m \})^{2n} \sum_{i=2}^m c_i^2 = (\max \{ \lambda_2, -\lambda_m \})^{2n} \chi^2(\rho^{(0)} \mid\mid \pi).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7b82e2d3cefcede8d4a7a72b1fa3ceea_l3.png "Rendered by QuickLaTeX.com")
![\[\tau_{\rm mix} \coloneqq \min \left\{ n \ge 1 : \max_{\rho^{(0)}} \norm{\rho^{(n)} - \pi}_{\rm TV} \le \frac{1}{2e} \right\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e491d914ce283944ca10dc9d96bdb1a5_l3.png "Rendered by QuickLaTeX.com")
 of the chain at time
of the chain at time  is the
is the ![\[\norm{\rho^{(n)} - \pi}_{\rm TV} \le e^{-\lfloor n / \tau_{\rm mix}\rfloor}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8a28da28e02550f209ce8ec1c8b5b79e_l3.png "Rendered by QuickLaTeX.com")
 , then
, then  .
. .
. such that (A) each process is individually a Markov chain with transition matrix
such that (A) each process is individually a Markov chain with transition matrix ![\[\prob\{x_{n+1} = j \mid x_n = i \} = \prob\{y_{n+1} = j \mid y_n = i \} = P_{ij}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8a91e29baa6fd219723bc325c68d9b75_l3.png "Rendered by QuickLaTeX.com")
 , then
, then  .
. and
and  is initialized in the stationary distribution
is initialized in the stationary distribution  . Let
. Let  be the time at which
be the time at which ![\[\tau_{\rho^{(0)}} \coloneqq \min\{ n \ge 0 : x_n = y_n\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-68580815d8347111c8a35c6767c24d3e_l3.png "Rendered by QuickLaTeX.com")
 control the convergence of the Markov chain
control the convergence of the Markov chain  .
.![\tau_{\rm mix} \le 2e \max_{\rho^{(0)}} \expect[\tau_{\rho^{(0)}}]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6bff20b3e6944c95a1e8bf2d625c22d0_l3.png "Rendered by QuickLaTeX.com") .
.![\[\norm{\rho^{(n)} - \pi}_{\rm TV} \le \prob\{\tau_{\rho^{(0)}} > n\}\le \frac{\expect[\tau_{\rho^{(0)}}]}{n}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-74fd53f9c863d5faf2edb9698e58854f_l3.png "Rendered by QuickLaTeX.com")
 . Then the left-hand side is at most
. Then the left-hand side is at most  , so
, so![\[\frac{1}{2e} \le \max_{\rho^{(0)}}\norm{\rho^{(\tau_{\rm mix})} - \pi}_{\rm TV} = \prob\{\tau_{\rho^{(0)}} > n\} \le \frac{\max_{\rho^{(0)}}\expect[\tau_{\rho^{(0)}}]}{\tau_{\rm mix}}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-61cf831f4db72e776d3122e3e323b7c1_l3.png "Rendered by QuickLaTeX.com")
 ). Therefore, one can also think of this Markov chain as a very inefficient way of generating a string of random bits.
). Therefore, one can also think of this Markov chain as a very inefficient way of generating a string of random bits. and a (uniform) random bit
and a (uniform) random bit  . Set
. Set  by changing the
by changing the  th bit of
th bit of  , and set
, and set  by changing the
by changing the  to
to  have been chosen by time
have been chosen by time  to be the time at which all positions
to be the time at which all positions ![\[\tau_{\rho^{(0)}} \le \tau_{\rm pick}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-878ab3b502681c3fb7c0453599af3293_l3.png "Rendered by QuickLaTeX.com")
![\expect[\tau_{\rm pick}]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ff405a11db183f95baecf7299661c685_l3.png "Rendered by QuickLaTeX.com") , it can be helpful to break into easier pieces. Let’s start with the simplest possible question: How many picks do we need to collect the first coupon? Just one. We always get a coupon in our first box.
, it can be helpful to break into easier pieces. Let’s start with the simplest possible question: How many picks do we need to collect the first coupon? Just one. We always get a coupon in our first box. of them are different from the first. So the probability of picking a new coupon in the second box is
of them are different from the first. So the probability of picking a new coupon in the second box is  . In fact,
. In fact, , and its mean is
, and its mean is  . On average, we need
. On average, we need  picks to collect the second coupon.
picks to collect the second coupon. coupons we haven’t collected and
coupons we haven’t collected and  . Thus, the number of picks is a geometric random variable with success probability
. Thus, the number of picks is a geometric random variable with success probability  .
. picks to pick the
picks to pick the  , we obtain that the total number of picks required is to collect all the coupons is
, we obtain that the total number of picks required is to collect all the coupons is![\[\expect[\tau_{\rm pick}] = 1 + \frac{N}{N-1} + \frac{N}{N-2} + \cdots + \frac{N}{N-(N-1)} = N \left( \frac{1}{N} + \frac{1}{N-1} + \cdots + \frac{1}{2} + 1 \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-db166dbcfe9198024b7dafb5de81fdf7_l3.png "Rendered by QuickLaTeX.com")
 denote the
denote the ![\[H_N = 1 + \frac{1}{2} + \cdots + \frac{1}{N}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-280c58a265450ae987b6c6236999bfd0_l3.png "Rendered by QuickLaTeX.com")
![\expect[\tau_{\rm pick}] = NH_N](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ec1cac0ce484b5e1ceef29a198523670_l3.png "Rendered by QuickLaTeX.com") . Since the
. Since the  , we have
, we have![\[\expect[\tau_{\rm pick}] \le N \log (N) + N.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2626a01a852627593ced1766177dcbbc_l3.png "Rendered by QuickLaTeX.com")
![\[\tau_{\rm mix} \le 2e \, \expect[\tau_{\rho^{(0)}}] \le 2e \, \expect[\tau_{\rm pick}] \le 2e \, N \log(N) + 2e \, N.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-73215e6c39a6bd7162aba749cd61e1bf_l3.png "Rendered by QuickLaTeX.com")
 .
.
![\[\prob\{\tau_{\rm pick} > N \log N + cN\} \le e^{-c}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-73f8883bcb323760364995473bcc3831_l3.png "Rendered by QuickLaTeX.com")
 and
and  , from which it follows that
, from which it follows that![\[\norm{\rho^{(n)} - \pi}_{\rm TV} \le \varepsilon \quad \text{provided that} \quad n \ge N \log N + N \log(1/\varepsilon).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-14c0bb8d63f118fc70a87253f64236af_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{\rho^{(n)} - \pi}_{\rm TV} \le \varepsilon \quad \text{provided that} \quad n \ge \frac{1}{2} N \log N + c \,N \log(1/\varepsilon)\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ce2a7b130eee9537cf737b90814aa4d3_l3.png "Rendered by QuickLaTeX.com")
 . The constant
. The constant  in this inequality cannot be improved. See section 5.3.1 of
in this inequality cannot be improved. See section 5.3.1 of  . When initialized from any starting distribution
. When initialized from any starting distribution  if all of its entries are strictly positive. Recall that a Markov chain with transition matrix
if all of its entries are strictly positive. Recall that a Markov chain with transition matrix  . For this post, all Markov chains will have state space
. For this post, all Markov chains will have state space  and
and ![\[\norm{\rho - \sigma}_{\rm TV} = \max_{S \subseteq \{1,\ldots,m\}} |\rho(S) - \sigma(S)| = \frac{1}{2} \sum_{i=1}^m \left| \rho_i - \sigma_i \right|.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7dd0b0e121d45408883369794becedff_l3.png "Rendered by QuickLaTeX.com")
 .
. . Thus, despite these distributions seeming quite similar to us, the total variation distance between
. Thus, despite these distributions seeming quite similar to us, the total variation distance between  , if
, if![\[\prob \{x = i\} = \rho_i \quad \text{for $i=1,2,\ldots,m$}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3537663be586f21278e7e32b7502af94_l3.png "Rendered by QuickLaTeX.com")
 on
on  is a distribution on
is a distribution on  such that if a pair of random variables
such that if a pair of random variables  is drawn from
is drawn from  . Denote the set of all couplings of
. Denote the set of all couplings of  .
.![\[\norm{\rho - \sigma}_{\rm TV} = \min_{\gamma \in \operatorname{Couplings}(\rho,\sigma)} \prob_{(x,y) \sim \gamma} \{ x \ne y \}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-918278c5556c178aee214db808000551_l3.png "Rendered by QuickLaTeX.com")
 represents the probability for variables
represents the probability for variables  drawn from joint distribution
drawn from joint distribution  . Then the coupling lemma tells us that there is a coupling
. Then the coupling lemma tells us that there is a coupling  . Indeed, such a coupling can be obtained by drawing
. Indeed, such a coupling can be obtained by drawing  . This defines a joint distribution
. This defines a joint distribution  with 100% probability.
with 100% probability. ,
, ![\[\norm{\rho - \sigma}_{\rm TV} \le \prob \{x \ne y \}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2b881043ba005f53ae84ada8ebe53191_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{\rho - \sigma}_{\rm TV} = \prob \{x \ne y\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e692d3e9715ed9b325be70775ba0eac3_l3.png "Rendered by QuickLaTeX.com")
 as
as  at an exponential rate.
at an exponential rate. is non-increasing
is non-increasing ![\[\norm{\rho^{(n+1)} - \pi}_{\rm TV} \le \norm{\rho^{(n)} - \pi}_{\rm TV} \quad \text{for every } n =0,1,2\ldots. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-49f8b3e1681a778cc0540ab25def9d1c_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{\rho^{(n)} - \pi}_{\rm TV} = \prob \{x_n\ne y_n\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8e78bf69e3ca31ba02e37fa3a64e5f52_l3.png "Rendered by QuickLaTeX.com")
 .
. , then run the two chains
, then run the two chains ![\[\prob\{x_{n+1}\ne y_{n+1}\} \le \prob\{x_n \ne y_n\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3904173ffb700f39526fb117f5a6c248_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{\rho^{(n+1)} - \pi}_{\rm TV} \le \prob\{x_{n+1}\ne y_{n+1}\} \le \prob\{x_n \ne y_n\} = \norm{\rho^{(n)} - \pi}_{\rm TV}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2409e83e870c68fe8d7f8e1a94903325_l3.png "Rendered by QuickLaTeX.com")
 .
. more carefully. Write it as
more carefully. Write it as
 , break into cases for all possible values
, break into cases for all possible values  for
for  to take
to take 
 be the minimum probability of moving between any two states
be the minimum probability of moving between any two states![\[p_{\rm min} \coloneqq \min_{1\le i,j\le m} P_{ij}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ca934d28f674adb9b8bc14ef320dd327_l3.png "Rendered by QuickLaTeX.com")
 to
to 
![\[\prob \{x_{n+1} \ne y_{n+1}\} \le (1 - p_{\rm min})\prob \{x_n \ne y_n\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-69b8b6e06ad981139c7c6ffd1ae6d1ba_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{\rho^{(n+1)}-\pi}_{\rm TV} \le \prob \{x_{n+1} \ne y_{n+1}\} \le (1 - p_{\rm min})\prob \{x_n \ne y_n\} = (1 - p_{\rm min}) \norm{\rho^{(n)} - \pi}_{\rm TV}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ce7570327a59ec2ede1a579d7f13564d_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{\rho^{(n)} - \pi}_{\rm TV} \le (1 - p_{\rm min})^n \norm{\rho^{(0)} - \pi}_{\rm TV} \le (1 - p_{\rm min})^n.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9119337225ce34aa99582b2b53069300_l3.png "Rendered by QuickLaTeX.com")
 . Fortunately, together with our earlier observation that distance to stationarity is non-increasing, we can upgrade this proof into a proof for the general case.
. Fortunately, together with our earlier observation that distance to stationarity is non-increasing, we can upgrade this proof into a proof for the general case. for which
for which  . Construct an auxilliary Markov chain
. Construct an auxilliary Markov chain  such that one step of the auxilliary chain consists of running
such that one step of the auxilliary chain consists of running ![\[z_0 = x_0, \:z_1 = x_{n_0}, \:z_2 = x_{2n_0},\ldots.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e5c67b34610d3cf8834e42621fd85218_l3.png "Rendered by QuickLaTeX.com")
 is
is  , we have
, we have![\[\norm{\rho^{(k\cdot n_0)} - \pi}_{\rm TV} \le (1-\delta)^k \norm{\rho^{(0)} - \pi}_{\rm TV} \le (1-\delta)^k \quad \text{for }k=0,1,2,\ldots,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f00ec64a989f2c387244cc13ca79cb63_l3.png "Rendered by QuickLaTeX.com")
 . Thus, since distance to stationarity is non-increasing, we have
. Thus, since distance to stationarity is non-increasing, we have![\[\norm{\rho^{(n)} - \pi}_{\rm TV} \le \norm{\rho^{(n_0 \cdot \lfloor n/n_0\rfloor)} - \pi}_{\rm TV} \le (1-\delta)^{\lfloor n/n_0\rfloor} \norm{\rho^{(0)} - \pi}_{\rm TV} \le (1-\delta)^{\lfloor n/n_0\rfloor}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c8c82df8e81ce5b1ca4fcbae23a11f4d_l3.png "Rendered by QuickLaTeX.com")
![\[\tau_{\rm mix} \coloneqq \min \left\{ n \ge 1 : \max_{\rho^{(0)}} \norm{\rho^{(n)} - \pi}_{\rm TV} \le \frac{1}{2e} \right\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ccf82253d6d08b59e651ef0c81a44e57_l3.png "Rendered by QuickLaTeX.com")
 steps:
steps: , then
, then  be the expected number of times the chain hits
be the expected number of times the chain hits  . Because the chain is primitive, all of the
. Because the chain is primitive, all of the ![\[\pi_i = \frac{a_i}{\sum_{j=1}^m a_j}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6abd189895ccf9707e3178558a0b5a2c_l3.png "Rendered by QuickLaTeX.com")
![\[a^\top P = a^\top. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d6885d4de71a8b8655bd542c1434c775_l3.png "Rendered by QuickLaTeX.com")
 denote the values of the chain and
denote the values of the chain and  denote the time at which the chain returns to
denote the time at which the chain returns to ![\[a_j = \sum_{n=1}^\infty \prob\{x_n = j, n_{\rm ret} > n\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-713cac4a8c97033b1cba87fe2632d46d_l3.png "Rendered by QuickLaTeX.com")
![\[a_j = \prob\{x_1 = j\} + \sum_{n=2}^\infty \prob\{x_n = j, n_{\rm ret} > n\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1f62dfb50726f4884c2cb790970d1a75_l3.png "Rendered by QuickLaTeX.com")
 . For the second term, break into cases depending on the value of the chain at time
. For the second term, break into cases depending on the value of the chain at time 
![\[a_j = P_{ij} + \sum_{n=2}^\infty \sum_{k\ne i} \prob\{x_{n-1} = k, n_{\rm ret} > n-1 \} P_{kj}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f243b52714b96a419b94980a12f078ee_l3.png "Rendered by QuickLaTeX.com")
 to
to  and exchange the order of summation to obtain
and exchange the order of summation to obtain![\[a_j = P_{ij} + \sum_{k\ne i} \left(\sum_{n=1}^\infty \prob\{x_n = k, n_{\rm ret} > n \}\right) P_{kj}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-cad1677f621e66a91f850ab352cedce0_l3.png "Rendered by QuickLaTeX.com")
 . Thus, since
. Thus, since  , we have
, we have![\[a_j = a_iP_{ij} + \sum_{k\ne i} a_k P_{kj} = \sum_{k=1}^m a_k P_{kj},\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-cecaa5c06b9ce9d9c3f4aa814dfc2323_l3.png "Rendered by QuickLaTeX.com")
 be a matrix and
be a matrix and  measured in some
measured in some  ? How do these answers change with different norms?
? How do these answers change with different norms? get except for some small failure probability
get except for some small failure probability  ?
?![\[\mathbb{E} \left\|B-X\right\|_{\rm F}^2 \le \left( 1 + \frac{k}{k-(r+1)}\right) \left\|B - \lowrank{B}_r \right\|_{\rm F}^2 \quad \text{for every $r \le k-2$}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6759ab11982f765f19dc615fb8e1fbdc_l3.png "Rendered by QuickLaTeX.com")
 is the
is the  where
where  , where
, where  denotes the
denotes the ![\[B\approx X \coloneqq QC.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-dbf0ef5be3e2a7fc39d609f4621d2dbd_l3.png "Rendered by QuickLaTeX.com")
 into a
into a  , as we did as steps 4 and 5 in the previous post.
, as we did as steps 4 and 5 in the previous post.![\[X = QC = QQ^*B = \Pi_{B\Omega} B.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0d1ae78fbe7d6d4d91b567795bd91f57_l3.png "Rendered by QuickLaTeX.com")
 denotes the
denotes the  . We call
. We call  the projection formula for the randomized SVD.
the projection formula for the randomized SVD. is
is ![\[U^*U = UU^* = I.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-33fedf7ef9e12832ea6188011e3874bd_l3.png "Rendered by QuickLaTeX.com")
 on matrices is
on matrices is ![\[\left\|UBV\right\|_{\rm UI} = \left\|B\right\|_{\rm UI} \quad \text{for all unitary matrices $U$, $V$ and any matrix $B$}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5fd16f43277f7e25ee2f910665984e09_l3.png "Rendered by QuickLaTeX.com")
 is said to be quadratic if there exists another unitarily invariant norm
is said to be quadratic if there exists another unitarily invariant norm ![\[\left\|B\right\|_{\rm Q}^2 = \left\|B^*B\right\|_{\rm UI} = \left\|BB^*\right\|_{\rm UI} \quad \text{for every matrix $B$.} \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-aac3c6aec691fb438adf6690682d855d_l3.png "Rendered by QuickLaTeX.com")
![\[\left\|B\right\|_{S_p}^p \coloneqq \sum_i \sigma_i(B)^p.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d032386a33d5df74c8a44235dafac359_l3.png "Rendered by QuickLaTeX.com")
 denote the decreasingly order
denote the decreasingly order  -norm
-norm  is the
is the ![\[\norm{B} = \left\|B\right\|_{S_\infty} \coloneqq \max_i \sigma_i(B) = \sigma_1(B).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-584051e6c083d9b865ffe52d79186a00_l3.png "Rendered by QuickLaTeX.com")
 . However, the Schatten
. However, the Schatten  since
since![\[\left\|B\right\|_{S_p}^2 = \left\|B^*B\right\|_{S_{p/2}}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-27e888ecff0b0c38df6e73a803f8aa1d_l3.png "Rendered by QuickLaTeX.com")
![\[\left\|B - X\right\|_{\rm Q}^2 \le \left\|B - \lowrank{B}_r\right\|_{\rm Q}^2 + \left\|\lowrank{B}_r - \Pi_{B\Omega} \lowrank{B}_r\right\|_{\rm Q}^2. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c1738a10a5c46d5455b8e2d5992ce98d_l3.png "Rendered by QuickLaTeX.com")
 to be an optimal rank-
to be an optimal rank- approximation to
approximation to ![\[\left\|B - \lowrank{B\right\|_r}_{\rm Q} \le \left\|B - C\right\|_{\rm Q} \quad \text{for any rank-$r$ matrix $C$}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-46106e1ebeaa349721ea4e81d171f70b_l3.png "Rendered by QuickLaTeX.com")
![\[\left\|B - X\right\|_{\rm Q}^2 - \left\|B - \lowrank{B}_r\right\|_{\rm Q}^2 \le \left\|\lowrank{B}_r - \Pi_{B\Omega} \lowrank{B}_r\right\|_{\rm Q}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e44e6de74ec072bf3d6c944ea6de77db_l3.png "Rendered by QuickLaTeX.com")

 and
and  , and the third is the triangle inequality. For the fifth line, we use the fact that
, and the third is the triangle inequality. For the fifth line, we use the fact that  is an orthoprojector and thus has unit spectral norm
is an orthoprojector and thus has unit spectral norm  together with the fact that
together with the fact that![\[\left\|BCD\right\|_{\rm UI} \le \norm{B} \cdot \left\|C\right\|_{\rm UI}\cdot \norm{D} \quad \text{for all matrices $B,C,D$.}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e329f332e4305af288668bfdf1ab4bf5_l3.png "Rendered by QuickLaTeX.com")
![\[(B - \lowrank{B}_r)(B - \lowrank{B}_r)^* = BB^* - \lowrank{B}_r\lowrank{B}_r^*.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9db3f7e6eb57261b556783a0a7d62fd0_l3.png "Rendered by QuickLaTeX.com")
![\[B = \onebytwo{U_1}{U_2} \twobytwo{\Sigma_1}{0}{0}{\Sigma_2}\onebytwo{V_1}{V_2}^*\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-54a749c18b5a76a0f468adee8b5fa58f_l3.png "Rendered by QuickLaTeX.com")
 and
and  both have orthonormal columns,
both have orthonormal columns, and
and  are square diagonal matrices with nonnegative entries,
are square diagonal matrices with nonnegative entries, and
and  have
have  matrix.
matrix. . Define
. Define![\[\twobyone{\Omega_1}{\Omega_2} \coloneqq \twobyone{V_1^*}{V_2^*} \Omega.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f620ec79bd8a3f6022efb5ca07b0d824_l3.png "Rendered by QuickLaTeX.com")
 is full-rank (i.e.,
is full-rank (i.e.,  ).
).
 so
so![\[\norm{(I-\Pi_{B\Omega})U_1\Sigma_1}_{\rm Q}^2 \le \norm{(I-\Pi_{B\Omega\Omega_1^\dagger})U_1\Sigma_1}_{\rm Q}^2 = \norm{\Sigma_1U_1^*(I-\Pi_{B\Omega\Omega_1^\dagger})U_1\Sigma_1}_{\rm UI}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2fdd85edd3e561ae5496031960d8901a_l3.png "Rendered by QuickLaTeX.com")
![\[\Sigma_1U_1^*(I-\Pi_{B\Omega\Omega_1^\dagger})U_1\Sigma_1.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f21f88902abb8532d54b098fa358ddc0_l3.png "Rendered by QuickLaTeX.com")
![\[B\Omega\Omega_1^\dagger = B\onebytwo{V_1}{V_2}\twobyone{\Omega_1}{\Omega_2}\Omega_1^\dagger = \onebytwo{U_1}{U_2} \twobytwo{\Sigma_1}{0}{0}{\Sigma_2} \twobyone{I}{\Omega_2\Omega_1^\dagger} = U_1\Sigma_1 + U_2\Sigma_2\Omega_2\Omega_1^\dagger.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e1dbcff75c12101b6274492896899b52_l3.png "Rendered by QuickLaTeX.com")
 takes the form
takes the form![\begin{align*}\Pi_{B\Omega\Omega_1^\dagger} &= (B\Omega\Omega_1^\dagger) \left[(B\Omega\Omega_1^\dagger)^*(B\Omega\Omega_1^\dagger)\right]^\dagger (B\Omega\Omega_1^\dagger)^* \\&= (U_1\Sigma_1 + U_2\Sigma_2\Omega_2\Omega_1^\dagger) \left[\Sigma_1^2 + (\Sigma_2\Omega_2\Omega_1^\dagger)^*(\Sigma_2\Omega_2\Omega_1^\dagger)\right]^\dagger (U_1\Sigma_1 + U_2\Sigma_2\Omega_2\Omega_1^\dagger)^*.\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d9e0cd585357787c460f6471f52ecb0a_l3.png "Rendered by QuickLaTeX.com")
 denotes the
denotes the ![\[\Sigma_1U_1^*(I-\Pi_{B\Omega\Omega_1^\dagger})U_1\Sigma_1 = \Sigma_1^2 - \Sigma_1^2 \left[\Sigma_1^2 + (\Sigma_2\Omega_2\Omega_1^\dagger)^*(\Sigma_2\Omega_2\Omega_1^\dagger)\right]^\dagger\Sigma_1^2. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b7dfa3f3cbcda5380b6b7ec0132fca2a_l3.png "Rendered by QuickLaTeX.com")
 is defined as
is defined as![\[A : H \coloneqq A - A(A+H)^\dagger A. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0ca6dd2810aa560e0d59d808c9b40bc8_l3.png "Rendered by QuickLaTeX.com")
![\[\Sigma_1U_1^*(I-\Pi_{B\Omega\Omega_1^\dagger})U_1\Sigma_1 = \Sigma_1^2 : [(\Sigma_2\Omega_2\Omega_1^\dagger)^*(\Sigma_2\Omega_2\Omega_1^\dagger)].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-dc620e38c4fb8a7855e330183c8313c9_l3.png "Rendered by QuickLaTeX.com")
![\[\left\|B - X\right\|_{\rm Q}^2\le \left\| \Sigma_2 \right\|_{\rm Q}^2 + \left\|\Sigma_1^2 : [(\Sigma_2\Omega_2\Omega_1^\dagger)^*(\Sigma_2\Omega_2\Omega_1^\dagger)]\right\|_{\rm UI}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ff8a161c38344c97e0d615d216d4555d_l3.png "Rendered by QuickLaTeX.com")
 . The
. The  is given by
is given by ![\[v = a \cdot \mathrm{curr}_a.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-50ff6a1f099e535f6dfd8525f45b4741_l3.png "Rendered by QuickLaTeX.com")
 is then
is then![\[v = h \cdot \mathrm{curr}_h.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-baea64abba5be2425a04d7d2316f7153_l3.png "Rendered by QuickLaTeX.com")
![\[\mathrm{curr}_{\rm total} = \mathrm{curr}_a + \mathrm{curr}_h = \frac{v}{a} + \frac{v}{h} = v \left(a^{-1}+h^{-1}\right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2ce6cd66280641977f8eea4d3d9f6cab_l3.png "Rendered by QuickLaTeX.com")
![\[a:h \coloneqq \frac{v}{\mathrm{curr}_{\rm total}} = \left( a^{-1}+h^{-1}\right)^{-1}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-46eba66bdf328c974e763d8c43422e75_l3.png "Rendered by QuickLaTeX.com")
 the parallel sum of
the parallel sum of 
 can be extended to all nonnegative numbers
can be extended to all nonnegative numbers ![\[a:h \coloneqq \lim_{b\downarrow a, \: k\downarrow h} b:k = \begin{cases}\left( a^{-1}+h^{-1}\right)^{-1}, & a,h > 0 ,\\0, & \textrm{otherwise}.\end{cases}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7c49cbcbcd8d8f75fa4f18a40562e718_l3.png "Rendered by QuickLaTeX.com")
![\[A:H = (A^{-1}+H^{-1})^{-1} \quad \text{for $A,H$ positive definite.}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-95cd7707ba37e3436926c73895d537bb_l3.png "Rendered by QuickLaTeX.com")
![\[a:h = \frac{1}{a^{-1}+h^{-1}} = \frac{ah}{a+h} = \frac{a(a+h)-a^2}{a+h} = a - a(a+h)^{-1}a.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e4e355840ccdeb857cec59c1173cca5b_l3.png "Rendered by QuickLaTeX.com")
![\[A : H = A - A(A+H)^\dagger A.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-85f7337339efbd593339ed3a5ff7e547_l3.png "Rendered by QuickLaTeX.com")
 ,
, for positive definite matrices,
for positive definite matrices, and
and  .
. is
is  is
is  ,
,  .
. .
. be the unitarily invariant norm
be the unitarily invariant norm ![\begin{align*}\left\|A:H\right\|_{\rm UI} &= \max_{M\succeq 0,\: \left\|M\right\|_{\rm UI}'\le 1} \tr((A:H)M) \\&= \max_{M\succeq 0,\: \left\|M\right\|_{\rm UI}'\le 1} \tr(M^{1/2}(A:H)M^{1/2})\\&= \max_{M\succeq 0,\: \left\|M\right\|_{\rm UI}'\le 1} \tr((M^{1/2}AM^{1/2}):(M^{1/2}HM^{1/2})) \\&\le \max_{M\succeq 0,\: \left\|M\right\|_{\rm UI}'\le 1} \left[\tr(M^{1/2}AM^{1/2}):\tr(M^{1/2}HM^{1/2}) \right] \\&\le \left[\max_{M\succeq 0,\: \left\|M\right\|_{\rm UI}'\le 1} \tr(M^{1/2}AM^{1/2})\right]:\left[\max_{M\succeq 0,\: \left\|M\right\|_{\rm UI}'\le 1} \tr(M^{1/2}HM^{1/2})\right]\\&= \left\|A\right\|_{\rm UI} : \left\|H\right\|_{\rm UI}.\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4d5c2226a0ddf81d346da401ff4b53b9_l3.png "Rendered by QuickLaTeX.com")
 , the third line is property 6, the fourth line is property 7, the fifth line is property 4, and the sixth line is duality.
, the third line is property 6, the fourth line is property 7, the fifth line is property 4, and the sixth line is duality.![\begin{align*}\left\|B - X\right\|_{\rm Q}^2&\le \left\| \Sigma_2 \right\|_{\rm Q}^2 + \left\|\Sigma_1^2 : [(\Sigma_2\Omega_2\Omega_1^\dagger)^*(\Sigma_2\Omega_2\Omega_1^\dagger)]\right\|_{\rm UI} \\&\le \left\| \Sigma_2 \right\|_{\rm Q}^2 + \left\|\Sigma_1^2\right\|_{\rm UI} : \left\|[(\Sigma_2\Omega_2\Omega_1^\dagger)^*(\Sigma_2\Omega_2\Omega_1^\dagger)]\right\|_{\rm UI} \\&= \left\| \Sigma_2 \right\|_{\rm Q}^2 + \left\|\Sigma_1\right\|_{\rm Q}^2 : \left\|\Sigma_2\Omega_2\Omega_1^\dagger\right\|^2_{\rm Q}.\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-df07b0b9660af51129fd29552ad6c650_l3.png "Rendered by QuickLaTeX.com")
 , we obtain the following expression for the expected error of the randomized SVD
, we obtain the following expression for the expected error of the randomized SVD![\[\mathbb{E} \left\|B - X\right\|_{\rm F}^2\le \left\| B - \lowrank{B\right\|_r }_{\rm F}^2 + \mathbb{E}\left\|\Sigma_2\Omega_2\Omega_1^\dagger\right\|^2_{\rm F}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3f9645663663b82d968efe393fc4c158_l3.png "Rendered by QuickLaTeX.com")
 . Remarkably, this can be done in closed form.
. Remarkably, this can be done in closed form. and
and  be independent standard Gaussian random matrices with
be independent standard Gaussian random matrices with  ,
,  . Then
. Then![\[\mathbb{E} \left\|SG\Gamma^\dagger R\right\|_{\rm F}^2 = \frac{1}{k-(r+1)}\left\|S\right\|_{\rm F}^2\left\|R\right\|_{\rm F}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-83f5771c01f2d031fd755830d363cda0_l3.png "Rendered by QuickLaTeX.com")
 does not appear:
does not appear:![\[\mathbb{E} \left\|SGW\right\|_{\rm F}^2 = \mathbb{E} \sum_{ij} \left(\sum_{k\ell} s_{ik}g_{k\ell}w_{\ell j} \right)^2 = \mathbb{E} \sum_{ijk\ell} s_{ik}^2 w_{\ell j}^2 = \left\|S\right\|_{\rm F}^2\left\|W\right\|_{\rm F}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-dc219fd0dab2748965ecbf439c051db5_l3.png "Rendered by QuickLaTeX.com")
 are independent, mean-zero, and variance-one. Thus, applying this result conditionally on
are independent, mean-zero, and variance-one. Thus, applying this result conditionally on  , we get
, we get![\[\mathbb{E} \left\|SG\Gamma^\dagger R\right\|_{\rm F}^2 =\mathbb{E}\left[ \mathbb{E}\left[ \left\|SG\Gamma^\dagger R\right\|_{\rm F}^2 \,\middle|\, \Gamma\right]\right] =\left\|S\right\|_{\rm F}^2\cdot \mathbb{E} \left\|\Gamma^\dagger R\right\|_{\rm F}^2. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-31c52f341c1e50d4e2c2e8583f3a7737_l3.png "Rendered by QuickLaTeX.com")
 , we rewrite using the
, we rewrite using the ![\[\mathbb{E} \left\|\Gamma^\dagger R\right\|_{\rm F}^2 = \mathbb{E} \tr \left(\Gamma^\dagger RR^* \Gamma^{*\dagger} \right)= \tr \left(RR^* \mathbb{E}[\Gamma^{*\dagger}\Gamma^\dagger] \right) = \tr \left(RR^* \mathbb{E}[(\Gamma\Gamma^*)^{-1}] \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-79bd47bd70f2015a46c038c4f0d0e5f7_l3.png "Rendered by QuickLaTeX.com")
 is known as an
is known as an  . Thus, we obtain
. Thus, we obtain![\[\mathbb{E} \left\|\Gamma^\dagger R\right\|_{\rm F}^2 = \tr \left(RR^* \mathbb{E}[(\Gamma\Gamma^*)^{-1}] \right) = \frac{\tr \left(RR^* \right)}{k-(r+1)} = \frac{\left\|R\right\|_{\rm F}^2}{k-(r+1)}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e874bd74b5ef81c13856de05454ea647_l3.png "Rendered by QuickLaTeX.com")
![\[\mathbb{E} \left\|SG\Gamma^\dagger R\right\|_{\rm F}^2 =\left\|S\right\|_{\rm F}^2\cdot \mathbb{E} \left\|\Gamma^\dagger R\right\|_{\rm F}^2 = \frac{1}{k-(r+1)}\left\|S\right\|_{\rm F}^2\left\|R\right\|_{\rm F}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b2ac63d48bafba095409bc148beed39c_l3.png "Rendered by QuickLaTeX.com")
 are independent and standard Gaussian as well. Plugging the matrix expectation bound to (9) then completes the analysis
are independent and standard Gaussian as well. Plugging the matrix expectation bound to (9) then completes the analysis
 matrix
matrix ![\[B \approx X \coloneqq Q C.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c5eb6a2bd93f7c628f485bd473e8676c_l3.png "Rendered by QuickLaTeX.com")
 where
where  and
and  and
and  and
and  is
is  .
.![\[B \approx X = QC = U\Sigma V^*.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8f7c31a46a04e9f0447ddc9209f26458_l3.png "Rendered by QuickLaTeX.com")
 as estimates of the
as estimates of the  numbers of storage, whereas
numbers of storage, whereas  numbers of storage. As I detailed at length in my blog post on
numbers of storage. As I detailed at length in my blog post on  in roughly
in roughly  . For these use cases, we don’t need the “SVD” part of the randomized SVD.
. For these use cases, we don’t need the “SVD” part of the randomized SVD. to the matrix
to the matrix  is than
is than  .
.![\[\mathbb{E} \left\|B - X\right\|_{\rm F}^2 \le \min_{r \le k-2} \left( 1 + \frac{r}{k-(r+1)} \right) \left\|B - \lowrank{B}_r\right\|^2_{\rm F}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-aa4c5d7163bac127432706a279ae7210_l3.png "Rendered by QuickLaTeX.com")
 . In particular, choosing
. In particular, choosing  , we see that the randomized SVD error is at most
, we see that the randomized SVD error is at most  times the best rank-
times the best rank-![\[\mathbb{E} \left\|B - X\right\|_{\rm F}^2 \le (1+r) \left\|B - \lowrank{B}_r \right\|^2_{\rm F} \quad \text{for $k=r+2$}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-29753ca17ed5f7def9297cf52157afb8_l3.png "Rendered by QuickLaTeX.com")
 , we see that the randomized SVD has at most twice the error of the best rank-
, we see that the randomized SVD has at most twice the error of the best rank-![\[\mathbb{E} \left\|B - X\right\|_{\rm F}^2 \le 2 \left\|B - \lowrank{B}_r\right\|^2_{\rm F} \quad \text{for $k=2r+1$}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1c11cf4647747653a356178b65c3585f_l3.png "Rendered by QuickLaTeX.com")
 for the randomized SVD suffices. These results hold even for worst-case matrices. For nice matrices with steadily decaying singular values, the randomized SVD can perform even better than equations (2)–(3) would suggest.
for the randomized SVD suffices. These results hold even for worst-case matrices. For nice matrices with steadily decaying singular values, the randomized SVD can perform even better than equations (2)–(3) would suggest.![\[\mathbb{E} \left\|B - X\right\|^2 \le \min_{r \le k-2} \left( 1 + \frac{2r}{k-(r+1)} \right) \left(\left\|B - \lowrank{B}_r \right\|^2 + \frac{\mathrm{e}^2}{k-r} \left\|B - \lowrank{B}_r\right\|^2_{\rm F} \right). \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8ee6f467b55a9e949f3636deca2589a5_l3.png "Rendered by QuickLaTeX.com")
 .
.![\[\norm{B} = \sigma_1(B),\quad \left\|B\right\|_{\rm F}^2 = \sum_i \sigma_i(B)^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2d3f6deda657f9f0f863a05450571e23_l3.png "Rendered by QuickLaTeX.com")
![\[\mathbb{E} \left\|B - X\right\|^2 \le \min_{r \le k-2} \left( 1 + \frac{2r}{k-(r+1)} \right) \left(\sigma_{r+1}^2 + \frac{\mathrm{e}^2}{k-r} \sum_{i>r} \sigma_i^2 \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d4bb8b98ee0928e647c7cdbd91332fcc_l3.png "Rendered by QuickLaTeX.com")
 , this bound demonstrates that the randomized SVD incurs errors based on the entire tail of
, this bound demonstrates that the randomized SVD incurs errors based on the entire tail of  . The randomized SVD is much worse than the best rank-
. The randomized SVD is much worse than the best rank- . Powering has the effect of amplifying the large singular values of
. Powering has the effect of amplifying the large singular values of  we compute the block
we compute the block ![\[B\Omega, BB^*B\Omega, BB^*BB^*B\Omega,\ldots,B(B^*B)^q\Omega\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2584aa789abb8f623fbbb14e114a5461_l3.png "Rendered by QuickLaTeX.com")
![\[Y = \begin{bmatrix} B\Omega & BB^*B\Omega & BB^*BB^*B\Omega& \cdots & B(B^*B)^q\Omega\end{bmatrix}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4139403e46250e757c4b558a38b9fba7_l3.png "Rendered by QuickLaTeX.com")