If a polynomial function is trapped in a box, how much can it wiggle? This question is answered by Markov’s inequality, which states that for a degree- polynomial
polynomial  that maps
that maps ![[-1,1]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-634fe4705a526f221e312cffd3ceda93_l3.png "Rendered by QuickLaTeX.com") into , it holds that
into , it holds that
(1) ![\[\max_{x \in [-1,1]} |p'(x)| \le n^2. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-63dfe56d2bb34f3fc695f743fdf67b40_l3.png "Rendered by QuickLaTeX.com")
is trapped within a square box ![[-1,1] \times [-1,1]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1c563b2ce110b55706b82573366be7c8_l3.png "Rendered by QuickLaTeX.com") , the fastest it can wiggle—as measured by its first derivative—is the square of its degree.
, the fastest it can wiggle—as measured by its first derivative—is the square of its degree.
How tight is this inequality? Do polynomials we know and love come close to saturating it, or is this bound very loose for them? A first polynomial which is natural to investigate is the power  . This function maps into , and the maximum value of its derivative is
. This function maps into , and the maximum value of its derivative is
![\[\max_{x \in [-1,1]} |p'(x)| = \max_{x \in [-1,1]} |nx^{n-1}| = n\ll n^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-fac35ca5506dda4220ce3027702d9f37_l3.png "Rendered by QuickLaTeX.com")
To saturate the inequality, we need something wigglier. The heavyweight champions for polynomial wiggliness are the Chebyshev polynomials  , which are motivated and described at length in this previous post. For our purposes, what’s important is that Chebyshev polynomials really know how to wiggle. Just look at how much more rapidly the degree-7 Chebyshev polynomial (blue solid line) moves around than the degree-7 power
, which are motivated and described at length in this previous post. For our purposes, what’s important is that Chebyshev polynomials really know how to wiggle. Just look at how much more rapidly the degree-7 Chebyshev polynomial (blue solid line) moves around than the degree-7 power  (orange dashed line).
(orange dashed line).
In addition to seeming a lot more wiggly to the eye, the degree- Chebyshev polynomials have much larger derivatives, saturating Markov’s inequality:
![\[\max_{x \in [-1,1]} |T_n'(x)| = n^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a5f7b0314564d6e87a09ef03981df060_l3.png "Rendered by QuickLaTeX.com")
These two examples illustrate a good rule of thumb. The derivative of a polynomial which is “simple” or “power-like” will be of size about , whereas the derivative of a “clever” or “Chebyshev-like” polynomial will be much higher at about  .
.
The inequality (1) was proven by Andrey Markov. It is much less well-known than Andrey Markov’s famous inequality in probability theory. A generalization of Markov’s inequality (1) was proven by Andrey’s brother Vladimir Markov, who proved a bound on the  th derivative of any polynomial mapping into :
th derivative of any polynomial mapping into :
(2) ![\[\max_{x \in [-1,1]} |p^{(k)}(x)| \le \max_{x \in [-1,1]} |T_d^{(k)}| = \frac{n^2(n^2-1^2)\cdots(n^2-(k-1)^2)}{1\times 3 \times \cdots \times (2k-1)}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-625bc4245d3cc1d47a534e8d824d9a9a_l3.png "Rendered by QuickLaTeX.com")
 . The inequality (2) is often called the Markov brothers’ inequality, and this name is sometimes also attached to the special case (1)—to help distinguish it from the probabilistic Markov inequality.
. The inequality (2) is often called the Markov brothers’ inequality, and this name is sometimes also attached to the special case (1)—to help distinguish it from the probabilistic Markov inequality.
For the rest of this post, we will focus on the basic Markov inequality (1). This inequality is easily extended to polynomials trapped in a box ![[a,b] \times [c,d]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4c5eee6ce9a02aaea141b2b44fa5d05e_l3.png "Rendered by QuickLaTeX.com") of general sidelengths. Indeed, any polynomial mapping
of general sidelengths. Indeed, any polynomial mapping ![[a,b]\to[c,d]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2a7815349e537249459ea754a1bc3744_l3.png "Rendered by QuickLaTeX.com") can be transmuted to a polynomial
can be transmuted to a polynomial  mapping to by an affine change of variables:
mapping to by an affine change of variables:
![\[\tilde{p}(x) = -1 + 2\cdot \frac{p(\ell(x)) - c}{d-c} \quad \text{for } \ell(x) = a + (b-a) \cdot \frac{x+1}{2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3f013f343441b1f0556a72513ea33d9e_l3.png "Rendered by QuickLaTeX.com")
 maps to and, by the chain rule, the maximum value of its derivative is
maps to and, by the chain rule, the maximum value of its derivative is ![\[\max_{t \in [a,b]} |p'(t)| = \frac{d-c}{b-a} \cdot \max_{x \in [-1,1]} |\tilde p'(x)|.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-20e2a9e85c0f8c7703ec8843c61ddfa8_l3.png "Rendered by QuickLaTeX.com")
Markov’s inequality (general domain/codomain). Let
to
. Then
(3)
![\[\max_{t \in [a,b]} |p'(t)| \le \frac{d-c}{b-a} \cdot n^2. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-752b97244cedaa80b3c201ab0d27ac8c_l3.png "Rendered by QuickLaTeX.com")
What’s Markov’s inequality good for? A lot, actually. In this post, we’ll see one application of this inequality: proving polynomial inapproximability. There are lots of times in computational math where its valuable to approximate some function like  ,
,  , or
, or  by a polynomial. There are lots of techniques for producing polynomial approximations and understanding the rate of convergence of the best-possible polynomial approximation. But sometimes we just want a quick estimate of the form, say, “a polynomial needs to be of degree at least to approximate that function up to additive error
by a polynomial. There are lots of techniques for producing polynomial approximations and understanding the rate of convergence of the best-possible polynomial approximation. But sometimes we just want a quick estimate of the form, say, “a polynomial needs to be of degree at least to approximate that function up to additive error  “. That is, we seek lower bounds on the polynomial needed to approximate a given function to a specified level of accuracy.
“. That is, we seek lower bounds on the polynomial needed to approximate a given function to a specified level of accuracy.
The general Markov’s inequality (3) provides a direct way of doing this. Our treatment follows Chapter 5 of Faster Algorithms via Approximation Theory by Sushant Sachdeva and Nisheesh K Vishnoi. The argument will consist of two steps. First, we trap the function in a box. Then, we show it wiggles a lot (i.e., there is a point at which the derivative is large). Therefore, by Markov’s inequality, we conclude that the degree of the polynomial must be sufficiently large.
Let’s start with the function and ask the question:
What polynomial degree
?
We are interested in the case where the interval is large, so we assume  . To address this question, suppose that is a polynomial that satisfies
. To address this question, suppose that is a polynomial that satisfies
![\[|p(t) - \e^{-t}| \le 0.1 \quad \text{for all }t \in [0,b].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e47cf528e4b9076d406c0fdbb28dd879_l3.png "Rendered by QuickLaTeX.com")
in a box and show it wiggles.
- Trap it in a box. Since
 for positive
for positive  , it therefore must hold that
, it therefore must hold that  . Therefore, the polynomial is trapped in the box
. Therefore, the polynomial is trapped in the box ![[0,b] \times [-0.1,1.1]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1fc744b538cdc16ff0989123627d98fd_l3.png "Rendered by QuickLaTeX.com") .
. - Show it wiggles. The function
 decreases quite rapidly. At zero, it is
decreases quite rapidly. At zero, it is  and at two, it is
and at two, it is  . Therefore, since
. Therefore, since  is within of for all , it must hold that
is within of for all , it must hold that  is at least
is at least  and
and  is at most
is at most  . Therefore, by the intermediate value theorem, there is some
. Therefore, by the intermediate value theorem, there is some  between
between  and
and  for which
for which ![\[p'(t^*) = \frac{p(2) - p(0)}{2-0} \ge \frac{0.9 - 0.24}{2}=0.33.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-128d3240b1f7024150800ae2aea13002_l3.png "Rendered by QuickLaTeX.com")
We are ready for the conclusion. Apply the bullet point above and Markov’s inequality (3) to obtain
![\[0.33 \le p'(t^*) \le \max_{t \in [0,b]} |p'(t)| \le \frac{1.1-(-0.1)}{b-0} \cdot n^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5abd7df66a224f73f72aeca9680f0d92_l3.png "Rendered by QuickLaTeX.com")
![\[n \ge \sqrt{\frac{0.33}{1.2}} \cdot \sqrt{b} > 0.5 \sqrt{b}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0aa42a6b5c1734de97fe13b82c211538_l3.png "Rendered by QuickLaTeX.com")
 to approximate on . This rough estimate actually turns out to be pretty sharp. By using an appropriate polynomial approximation technique (say, interpolation at the Chebyshev points), a polynomial of degree
to approximate on . This rough estimate actually turns out to be pretty sharp. By using an appropriate polynomial approximation technique (say, interpolation at the Chebyshev points), a polynomial of degree  also suffices to approximate to this level of accuracy on .
also suffices to approximate to this level of accuracy on .
We’ve illustrated with just a single example, but this same technique can also be used to give (sometimes sharp, sometimes not) inapproximability results for other functions we know and love like  and
and  . For a bit of fun, see if you can get results for approximating a power
. For a bit of fun, see if you can get results for approximating a power  on by a polynomial of degree
on by a polynomial of degree  . You may be surprised by what you find!
. You may be surprised by what you find!
I find the Markov inequality technique for proving polynomial inapproximability to be pretty cool. As we saw in a previous post, we usually understand the difficulty of approximating a function in terms of the rate of convergence of the best (polynomial) approximation, which is tied to fine properties of the function and its smoothness. The Markov inequality approach answers a different question and uses entirely different information about the function. Rather than asking about the asymptotic rate of convergence, the Markov inequality approach tells you at what degree does a polynomial start approximating the function at all. And rather than using information about smoothness, the Markov approach shows that polynomial inapproximability is hard for any function that changes a lot over a small interval. As an exercise, you can show that the same argument for inapproximability of also shows a polynomial of degree  is necessary for approximating the ramp function
is necessary for approximating the ramp function
![\[f_{\rm ramp}(t) = \begin{cases} 1-t/2, & 0 \le t \le 2, \\ 0, & 2 < t \le b.\end{cases}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-26e453856802dfaca0eef8e4b383a649_l3.png "Rendered by QuickLaTeX.com")
converge at an exponential rate, whereas polynomial approximations to  converge at the rate
converge at the rate  . But in terms of the polynomial degree to start approximating these functions, both functions require the same degree
. But in terms of the polynomial degree to start approximating these functions, both functions require the same degree  . Pretty neat, I think.
. Pretty neat, I think.
Reference. This blog post is my take on an argument presented in Chapter 5 of Faster Algorithms via Approximation Theory by Sushant Sachdeva and Nisheesh K Vishnoi. It’s a very nice monograph, and I highly recommend you check it out!
 , but these results give poor results for intermediate block sizes
, but these results give poor results for intermediate block sizes  . But
. But  . Check out
. Check out ![\[K = \begin{bmatrix} G & AG & \cdots & A^{t-1}G \end{bmatrix},\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3a6ec37fe54d09f2ee9f6f2c648dafd1_l3.png "Rendered by QuickLaTeX.com")
 is an
is an  real
real  is an
is an  random
random ![\[p_a(u) = a_1 + a_2 u+ \cdots + a_t u^{t-1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ba898ff67858e9c5a76463593337b542_l3.png "Rendered by QuickLaTeX.com")
 and have written the polynomial
and have written the polynomial  parametrically in terms of its vector of coefficients
parametrically in terms of its vector of coefficients  . The polynomials of degree at most
. The polynomials of degree at most  provide a
provide a  to be complex numbers.
to be complex numbers.
 be
be  (distinct) input locations. If we evaluate
(distinct) input locations. If we evaluate  at each number, we obtain a list of (output) values, which we denote by
at each number, we obtain a list of (output) values, which we denote by  of
of ![\[p_a(\lambda_i) = \sum_{j=1}^t \lambda_i^{j-1} a_j.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-eaaf53acb1f30e1ced80af4704800e02_l3.png "Rendered by QuickLaTeX.com")
 are a nonlinear function of the input values
are a nonlinear function of the input values  but a linear function of the coefficients
but a linear function of the coefficients  . We may call the mapping
. We may call the mapping  the coefficients-to-values map.
the coefficients-to-values map.
 . It is given by the formula
. It is given by the formula ![\[V= \begin{bmatrix} 1 & \lambda_1 & \lambda_1^2 & \cdots & \lambda_1^{t-1} \\ 1 & \lambda_2 & \lambda_2^2 & \cdots & \lambda_2^{t-1} \\ 1 & \lambda_3 & \lambda_3^2 & \cdots & \lambda_3^{t-1} \\ \vdots & \vdots & \vdots & \ddots & \vdots \\ 1 & \lambda_s & \lambda_s^2 & \cdots & \lambda_s^{t-1} \end{bmatrix} \in \complex^{s\times t}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2d100f803f43a4910ba6c03d2edde384_l3.png "Rendered by QuickLaTeX.com")
 .
.
 so the number of locations
so the number of locations  equals the number of coefficients
equals the number of coefficients  . Its inverse
. Its inverse  maps a set of values
maps a set of values ![\[p_a(\lambda_i) = f_i \quad \text{for } i =1,\ldots,t .\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0cab3d96a2006c60b95e665b3c9afc97_l3.png "Rendered by QuickLaTeX.com")
 , obtaining a vector of coefficients
, obtaining a vector of coefficients  . Then, define the interpolating polynomial
. Then, define the interpolating polynomial ![\[q(u) = a_1 + a_2 u + \cdots + a_t u^{t-1} \quad \text{with } a = V^{-1} f. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b44af0dbb2f4e6b3c74a40b23d074f68_l3.png "Rendered by QuickLaTeX.com")
 interpolates the values
interpolates the values  ,
,  .
.
 that is zero at the locations
that is zero at the locations  but nonzero at the first location
but nonzero at the first location  . (Remember that we have assumed that
. (Remember that we have assumed that  are distinct.) Further, by rescaling this polynomial to
are distinct.) Further, by rescaling this polynomial to![\[\ell_1(u) = \frac{(u - \lambda_2)(u - \lambda_3)\cdots(u-\lambda_t)}{(\lambda_1 - \lambda_2)(\lambda_1 - \lambda_3)\cdots(\lambda_1-\lambda_t)},\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-174996c439ec13555388925748a3269e_l3.png "Rendered by QuickLaTeX.com")
 , we can define a similar polynomial
, we can define a similar polynomial ![\[\ell_i(u) = \frac{(u - \lambda_1)\cdots(u-\lambda_{i-1}) (u-\lambda_{i+1})\cdots(u-\lambda_t)}{(\lambda_i - \lambda_1)\cdots(\lambda_i-\lambda_{i-1}) (\lambda_i-\lambda_{i+1})\cdots(\lambda_i-\lambda_t)}, \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5453dea91bda2e744ca98b5dc0ae9790_l3.png "Rendered by QuickLaTeX.com")
 for
for  . Using the Dirac delta symbol, we may write
. Using the Dirac delta symbol, we may write ![\[\ell_i(\lambda_j) = \delta_{ij} = \begin{cases} 1, & i = j, \\ 0, & i\ne j. \end{cases}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-64c4effeb0f12225a76059d89ccce692_l3.png "Rendered by QuickLaTeX.com")
 are called the
are called the  . Below is an interactive illustration of the second Lagrange polynomial
. Below is an interactive illustration of the second Lagrange polynomial  associated with the points
associated with the points  (with
(with  ).
).
 and sum up, obtaining
and sum up, obtaining ![\[q(u) = \sum_{i=1}^t f_i \ell_i(u).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c1c5043718b5df795a883a6a60d26aaf_l3.png "Rendered by QuickLaTeX.com")
![\[q(\lambda_j) = \sum_{i=1}^t f_i \ell_i(\lambda_j) = \sum_{i=1}^t f_i \delta_{ij} = f_j. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-90032b377ebe250ef36dccba2186b6ce_l3.png "Rendered by QuickLaTeX.com")
 . To convert between these formulas, we just need to express the Lagrange polynomial basis in the monomial basis.
. To convert between these formulas, we just need to express the Lagrange polynomial basis in the monomial basis. , and consider the fourth unnormalized Lagrange polynomial
, and consider the fourth unnormalized Lagrange polynomial ![\[(u - \lambda_1) (u - \lambda_2) (u - \lambda_3).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d3fe2471c7c0557476992f6f7a1361cf_l3.png "Rendered by QuickLaTeX.com")
 , we obtain the expression
, we obtain the expression ![\[(u - \lambda_1) (u - \lambda_2) (u - \lambda_3) = u^3 - (\lambda_1 + \lambda_2 + \lambda_3) u^2 + (\lambda_1\lambda_2 + \lambda_1\lambda_3 + \lambda_2\lambda_3)u - \lambda_1\lambda_2\lambda_3.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ad4d01202c2b9a69e395412829cc7fed_l3.png "Rendered by QuickLaTeX.com")
 , we recognize some pretty distinctive expressions involving the
, we recognize some pretty distinctive expressions involving the 
 , the
, the  is defined as the sum of all products
is defined as the sum of all products  of
of ![\[e_k(\mu_1,\ldots,\mu_{t-1}) = \sum_{i_1 < i_2 < \cdots < i_k} \mu_{i_1}\mu_{i_2}\cdots \mu_{i_k}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-49b00bf8f1a0f6fc399e029516a4682e_l3.png "Rendered by QuickLaTeX.com")
 :
:![\[(u + \mu_1) (u + \mu_2) \cdots (u+\mu_k) = \sum_{j=0}^k e_{k-j}(\mu_1,\ldots,\mu_k) u^j.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-fe26dd7ba2e215fba4fea5cd28147cb2_l3.png "Rendered by QuickLaTeX.com")
 denote the list of locations without
denote the list of locations without ![\[\ell_i(u) = \frac{\sum_{j=1}^t e_{t-j}(-\lambda_{-i})u^{j-1}}{\prod_{k\ne i} (\lambda_i - \lambda_k)}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-15d6cb602dcf916e14363b25a013870d_l3.png "Rendered by QuickLaTeX.com")
![\[q(u) = \sum_{i=1}^t f_i\ell_i(u) = \sum_{i=1}^t \sum_{j=1}^t f_i \frac{e_{t-j}(-\lambda_{-i})}{\prod_{k\ne i} (\lambda_i - \lambda_k)} u^{j-1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-17012971c8949a309b08ae230bf7fa94_l3.png "Rendered by QuickLaTeX.com")
![\[q(u) = \sum_{j=1}^t \sum_{i=1}^t f_i \frac{e_{t-j}(-\lambda_{-i})}{\prod_{k\ne i} (\lambda_i - \lambda_k)} u^{j-1} = \sum_{j=1}^t \left(\sum_{i=1}^t \frac{e_{t-j}(-\lambda_{-i})}{\prod_{k\ne i} (\lambda_i - \lambda_k)}\cdot f_i \right) u^{j-1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-10960bce48c6a124c253db4c1272f2e2_l3.png "Rendered by QuickLaTeX.com")
![\[a_j = \sum_{i=1}^t \frac{e_{t-j}(-\lambda_{-i})}{\prod_{k\ne i} (\lambda_i - \lambda_k)}\cdot f_i.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2bb3d1280c99ca124084f031ed343679_l3.png "Rendered by QuickLaTeX.com")
![\[(V^{-1})_{ji} = \frac{e_{t-j}(-\lambda_{-i})}{\prod_{k\ne i} (\lambda_i - \lambda_k)}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c3d618b9ee7195d040d1875f76fb1d67_l3.png "Rendered by QuickLaTeX.com")
 , the condition number of
, the condition number of ![\[\kappa_{\uinorm{\cdot}}(V) = \uinorm{V} \uinorm{\smash{V^{-1}}}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-cf4da36f5f2ce121d2d2512423310221_l3.png "Rendered by QuickLaTeX.com")
 is chosen to be the (
is chosen to be the (![\[\norm{A}_1= \max_{x \ne 0} \frac{\norm{Ax}_1}{\norm{x}_1} \quad \text{where } \norm{x}_1 = \sum_i |x_i|.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-beb68f2e11bc5c6e30b06fec3b886ff3_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{A}_\infty = \max_j \sum_i |A_{ij}|. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-936a7309e99d6c03a128b3a7771c88df_l3.png "Rendered by QuickLaTeX.com")
 .
.
 is straightforward. Indeed, setting
is straightforward. Indeed, setting  and using (5), we compute
and using (5), we compute ![\[\norm{V}_1= \max_{1\le j \le t} \sum_{i=1}^t |\lambda_i|^{j-1} \le \max_{1\le j \le t} tM^{j-1} = t\max\{1,M^{t-1}\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ec5f488cb8233df69dd22a00e9bae784_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{V}_1\le t(1+M)^{t-1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8cecac332b3fcbe899304e963ec1fe47_l3.png "Rendered by QuickLaTeX.com")
 . Fortunately, we have already done most of the hard work needed to bound this quantity. Using our expression (4) for the entries of
. Fortunately, we have already done most of the hard work needed to bound this quantity. Using our expression (4) for the entries of ![\[\norm{\smash{V^{-1}}}_1= \max_{1\le j \le t} \sum_{i=1}^t |(V^{-1})_{ij}| = \max_{1\le j \le t}\frac{ \sum_{i=1}^t |e_{t-i}(-\lambda_{-j})|}{\prod_{k\ne j} |\lambda_j- \lambda_k|}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b431915bf4dd9e0a3087cd91f5e3533a_l3.png "Rendered by QuickLaTeX.com")
![\[|e_k(\mu_1,\ldots,\mu_s)| \le e_k(|\mu_1|,\ldots,|\mu_s|).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-73a7d10b6aadf0271cb538d648049f13_l3.png "Rendered by QuickLaTeX.com")
 , we obtain
, we obtain![\[\norm{\smash{V^{-1}}}_1\le \max_{1\le j \le t}\frac{ \sum_{i=1}^t e_{t-i}(|\lambda_{-j}|)}{\prod_{k\ne j} |\lambda_j- \lambda_k|}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-34c4b502a43c91513781e51c25190ad1_l3.png "Rendered by QuickLaTeX.com")
 to obtain the expression
to obtain the expression ![\[\norm{\smash{V^{-1}}}_1\le \max_{1\le j \le t}\frac{ \prod_{k\ne j} (1 + |\lambda_k|)}{\prod_{k\ne j} |\lambda_j- \lambda_k|}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9fee19675094e57026eaf8571e060317_l3.png "Rendered by QuickLaTeX.com")
 . To bound the denominator, let
. To bound the denominator, let  be the smallest distance between two locations. Using
be the smallest distance between two locations. Using  and
and  , we can weaken the bound (6) to obtain
, we can weaken the bound (6) to obtain ![\[\norm{\smash{V^{-1}}}_1\le \left(\frac{1+M}{\mathrm{gap}}\right)^{t-1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4df225e8dea0e69df68907fc81ab32e6_l3.png "Rendered by QuickLaTeX.com")
 from above, we obtain a bound on the condition number
from above, we obtain a bound on the condition number ![\[\kappa_1(V) \le t \left( \frac{(1+M)^2}{\mathrm{gap}} \right)^{t-1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a88bac288484194059179a7e854bc368_l3.png "Rendered by QuickLaTeX.com")
 . Then
. Then ![\[\norm{\smash{V^{-1}}}_1\le \left(\frac{1+M}{\mathrm{gap}}\right)^{t-1}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-933010efafd6e0336cdf5b4071c2c90b_l3.png "Rendered by QuickLaTeX.com")
 polynomial has precisely
polynomial has precisely  of the values
of the values  ? Well, if we set all the coefficients
? Well, if we set all the coefficients  to zero, then
to zero, then  as well. So to avoid this trivial case, we should enforce a normalization condition on the coefficient vector
as well. So to avoid this trivial case, we should enforce a normalization condition on the coefficient vector  . With this setting, we are ready to compute. Begin by observing that
. With this setting, we are ready to compute. Begin by observing that ![\[\min_{\norm{a}_1 = 1} \norm{Va}_1 = \min_{a\ne 0} \frac{\norm{Va}_1}{\norm{a}_1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3a21773d318eeb447cc3deec7d2ec9e1_l3.png "Rendered by QuickLaTeX.com")
 , obtaining
, obtaining ![\[\min_{\norm{a}_1 = 1} \norm{Va}_1 = \min_{a\ne 0} \frac{\norm{Va}_1}{\norm{a}_1} = \min_{f\ne 0} \frac{\norm{f}_1}{\norm{\smash{V^{-1}f}}_1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e223271ff5f99accd84933f4936864bd_l3.png "Rendered by QuickLaTeX.com")
![\[\left(\min_{\norm{a}_1 = 1} \norm{Va}_1\right)^{-1} = \max_{f\ne 0} \frac{\norm{\smash{V^{-1}f}}_1}{\norm{f}_1} = \norm{\smash{V^{-1}}}_1.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-745fdb308eb43cd71eb1fda9a7b16762_l3.png "Rendered by QuickLaTeX.com")
![\[\min_{\norm{a}_1 = 1} \norm{Va}_1 = \norm{\smash{V^{-1}}}_1^{-1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-145e10d104c261b8eba6886d9e5a88d0_l3.png "Rendered by QuickLaTeX.com")
![\[\min_{\uinorm{v} = 1} \uinorm{Av} = \min_{v\ne 0} \frac{\uinorm{Av}}{\uinorm{v}} = \uinorm{\smash{A^{-1}}}^{-1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c07d1dd4195814db2ab892e8587fb9b4_l3.png "Rendered by QuickLaTeX.com")
 on the values
on the values  and locations
and locations ![\[|p(\lambda_1)| + \cdots + |p(\lambda_t)| \ge \left(\frac{\mathrm{gap}}{1+M}\right)^{t-1} (|a_1| + \cdots + |a_t|).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f011798a0af29d677a21bf1d0fb00152_l3.png "Rendered by QuickLaTeX.com")
 be a
be a ![\expect[zf(z)] = \expect[f'(z)]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7dba10c0a29b0f68eefa135a0e67d0cb_l3.png "Rendered by QuickLaTeX.com") .
.![\expect[z^2]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b789f7d051bbf792fea37f44f88b1f83_l3.png "Rendered by QuickLaTeX.com") of a standard Gaussian random variable
of a standard Gaussian random variable  to obtain
to obtain ![\[\expect[z^2] = \expect[z f(z)] = \expect[f'(z)] = \expect[1] = 1.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-72017e2492468cdba39e4878b12f76aa_l3.png "Rendered by QuickLaTeX.com")
 , we compute
, we compute ![\[\expect[z^4] = \expect[zf(z)] = \expect[f'(z)] = 3\expect[z^2] = 3.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d7582a91dd80de04d4bb3bd17c2969cd_l3.png "Rendered by QuickLaTeX.com")
![\[\expect[z^{2p}] = \expect[z\cdot z^{2p-1}] = (2p-1) \expect[z^{2p-2}] = (2p-1)(2p-3) \expect[z^{2p-4}] = \cdots = (2p-1)(2p-3)\cdots 1.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-463cff05d7ce30d54b9325a0b4d32da8_l3.png "Rendered by QuickLaTeX.com")
 th moment of a standard Gaussian random variable is
th moment of a standard Gaussian random variable is  , where
, where  indicates the (in)famous
indicates the (in)famous  .
.
![\expect[|z|]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8dcb87553abe8c547e97479e4ef0ad22_l3.png "Rendered by QuickLaTeX.com") . To do so, we choose
. To do so, we choose  to be the
to be the ![\[\operatorname{sign}(x) = \begin{cases} 1, & x > 0, \\ 0 & x = 0, \\ -1 & x < 0.\end{cases}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-28ef3c2b8f17f4fc65f112fc1ea06c86_l3.png "Rendered by QuickLaTeX.com")
 , where
, where  denotes the famous
denotes the famous ![\[\expect[|z|] = \expect[zf(z)] = \expect[f'(z)] = 2\expect[\delta(z)].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ff50b28c78f8b8d69843835ac3bd7fab_l3.png "Rendered by QuickLaTeX.com")
![\expect[\delta(z)]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-078369af4f64b86ff6fe4d075710549d_l3.png "Rendered by QuickLaTeX.com") , write the integral out using the
, write the integral out using the  of the standard Gaussian distribution:
of the standard Gaussian distribution: ![\[\expect[|z|] = 2\expect[\delta(z)] = 2\int_{-\infty}^\infty \phi(x) \delta(x) \, \mathrm{d} x = 2\phi(0).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-96eedb8599b918aed9f53f71548b47d8_l3.png "Rendered by QuickLaTeX.com")
![\[\phi(x) = \frac{1}{\sqrt{2\pi}} \exp \left( - \frac{x^2}{2} \right),\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-92edc4a207255caae1eedb17381a807f_l3.png "Rendered by QuickLaTeX.com")
![\[\expect[|z|] = 2 \cdot \frac{1}{\sqrt{2 \pi}} = \sqrt{\frac{2}{\pi}}}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d1d32c2b5642230b0f179641bf6b5a65_l3.png "Rendered by QuickLaTeX.com")
 denote the eigenvalues of
denote the eigenvalues of  .
.  . After many iterations,
. After many iterations,  approaches an eigenvector of
approaches an eigenvector of  approaches an eigenvalue. Letting
approaches an eigenvalue. Letting  denote the initial vector, the
denote the initial vector, the  and the
and the ![\[\mu^{(t)} = \left(x^{(t)}\right)^\top Ax^{(t)}= \frac{\left(x^{(0)}\right)^\top A^{2t+1} x^{(0)}}{\left(x^{(0)}\right)^\top A^{2t} x^{(0)}}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8744da7cad08a86191f52369bd86e7b3_l3.png "Rendered by QuickLaTeX.com")
 of
of ![\[\mu^{(t)} = \frac{\sum_{i=1}^n \lambda_i^{2t+1} z_i^2}{\sum_{i=1}^n \lambda_i^{2t} z_i^2},\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-91c240175a5862804e47c506606be709_l3.png "Rendered by QuickLaTeX.com")
 as an approximation to the dominant eigenvalue
as an approximation to the dominant eigenvalue ![\[\frac{\lambda_1 - \mu^{(t)}}{\lambda_1} = \frac{\sum_{i=1}^n (\lambda_1 - \lambda_i)/\lambda_1 \cdot \lambda_i^{2t} z_i^2}{\sum_{i=1}^n \lambda_i^{2t} z_i^2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-667bb71dd8089d5b26d87d625b82e884_l3.png "Rendered by QuickLaTeX.com")
![\[\frac{\lambda_1 - \lambda_i}{\lambda_1}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0a300e8efbe358c2e744445cb4d0c552_l3.png "Rendered by QuickLaTeX.com")
 and is at most one for
and is at most one for  . Therefore, the error is bounded as
. Therefore, the error is bounded as ![\[\frac{\lambda_1 - \mu^{(t)}}{\lambda_1} \le \frac{\sum_{i=2}^n \lambda_i^{2t} z_i^2}{\lambda_1^{2t} z_1^2 + \sum_{i=2}^n \lambda_i^{2t} z_i^2} = \frac{c^2}{\lambda_1^{2t} z_1^2 + c^2} \quad \text{for } c^2 = \sum_{i=2}^n \lambda_i^{2t} z_i^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-752c7ced1eb1bd8cc0ddf1eacdbc5b1c_l3.png "Rendered by QuickLaTeX.com")
 to consolidate the terms in this expression not depending on
to consolidate the terms in this expression not depending on  . Since
. Since  with respect to only the randomness in the first Gaussian variable
with respect to only the randomness in the first Gaussian variable ![\[f'(x) = \frac{c^2}{\lambda_1^{2t}x^2 + c^2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5a451acfc5ab3bdec6db9fab5c32cf9b_l3.png "Rendered by QuickLaTeX.com")
![\[f(x) = \frac{c}{\lambda_1^t} \arctan \left( \frac{\lambda_1^t}{c} \cdot x \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6e54f3861046933630d2185bac2fb159_l3.png "Rendered by QuickLaTeX.com")
![\[\expect_{z_1}\left[\frac{\lambda_1 - \mu^{(t)}}{\lambda_1}\right] \le \expect_{z_1} \left[z_1 \cdot \frac{c}{\lambda_1} \arctan \left( \frac{\lambda_1^t}{c} \cdot z_1 \right) \right] = \expect_{z_1} \left[|z_1| \cdot \frac{c}{\lambda_1^t} \left|\arctan \left( \frac{\lambda_1^t}{c} \cdot z_1 \right)\right| \right].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b8e649fd7a63c495f6f2fa09d9534c3b_l3.png "Rendered by QuickLaTeX.com")
 , so we can bound
, so we can bound![\[\expect_{z_1}\left[\frac{\lambda_1 - \mu^{(t)}}{\lambda_1}\right] \le = \frac{\pi}{2} \cdot \frac{c}{\lambda_1^t} \cdot \expect_{z_1} \left[|z_1|\right] \le \sqrt{\frac{\pi}{2}} \cdot \frac{c}{\lambda_1^t}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e9d84cb3710fdc80b0166f5491e5b3da_l3.png "Rendered by QuickLaTeX.com")
![\expect[|z|] = \sqrt{2/\pi}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-72fae41f864e69e247552190c6b1ef2e_l3.png "Rendered by QuickLaTeX.com") for a standard Gaussian variable
for a standard Gaussian variable ![\[c = \sqrt{\sum_{i=2} \lambda_i^{2t} z_i^2} \le \lambda_2^{t} \sqrt{\sum_{i=2}^n z_i^2} = \lambda_2^t \norm{(z_2,\ldots,z_n)}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7facff60e6dd9b6579d16ed28a6b0f47_l3.png "Rendered by QuickLaTeX.com")
 times the length of a vector of
times the length of a vector of  standard Gaussian entries. As we’ve
standard Gaussian entries. As we’ve  . Thus,
. Thus, ![\expect[c] \le \lambda_2^t \sqrt{n-1}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-334812a1686dc037c03bd91572144ee7_l3.png "Rendered by QuickLaTeX.com") . We conclude that the expected error for power iteration is
. We conclude that the expected error for power iteration is ![\[\expect\left[\frac{\lambda_1 - \mu^{(t)}}{\lambda_1}\right] = \expect_c\left[\expect_{z_1} \left[ \frac{\lambda_1 - \mu^{(t)}}{\lambda_1} \right]\right] \le \sqrt{\frac{\pi}{2}} \cdot \expect\left[ \frac{c}{\lambda_1^t} \right] =\sqrt{\frac{\pi}{2}} \cdot \left(\frac{\lambda_2}{\lambda_1}\right)^t \cdot \sqrt{n-1} .\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-71e78c2a3b2280f94d13d08cfeed954c_l3.png "Rendered by QuickLaTeX.com")
 .
.
![\[\text{Find $x$ such that } Ax = b. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-29d9a4727a905120c7e471f5f6d88048_l3.png "Rendered by QuickLaTeX.com")
 . Beginning from an initial iterate
. Beginning from an initial iterate  , randomized Kaczmarz works as follows. For
, randomized Kaczmarz works as follows. For  :
:
 with probability
with probability  .
. holds exactly:
holds exactly: ![\[x_{t+1} \coloneqq x_t + \frac{b_{i_t} - a_{i_t}^\top x_t}{\norm{a_{i_t}}^2} a_{i_t}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-99cc99e6de2ca367d24ee6f20e610735_l3.png "Rendered by QuickLaTeX.com")
 denotes the
denotes the  th row of
th row of  should we use? The answer to this question may depend on whether the system (1) is
should we use? The answer to this question may depend on whether the system (1) is ![\[p_j = \frac{\norm{a_j}^2}{\norm{A}_{\rm F}^2} \quad \text{for } j = 1,2,\ldots,n. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c0282178b77487afd710ad66bb50707b_l3.png "Rendered by QuickLaTeX.com")
![\[p_j = \frac{1}{n} \quad \text{for } j = 1,2,\ldots,n. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b0a9325d8b121aaab8d4547fa378ffed_l3.png "Rendered by QuickLaTeX.com")
 with entries
with entries  , and choose the right-hand side
, and choose the right-hand side  with standard Gaussian random entries. The convergence of standard RK with sampling rule (1) and uniform RK with sampling rule (2) is shown in the plot below. After a million iterations, the difference in final accuracy is dramatic: the final relative error 0.00012 was uniform RK and 0.67 for standard RK!
with standard Gaussian random entries. The convergence of standard RK with sampling rule (1) and uniform RK with sampling rule (2) is shown in the plot below. After a million iterations, the difference in final accuracy is dramatic: the final relative error 0.00012 was uniform RK and 0.67 for standard RK!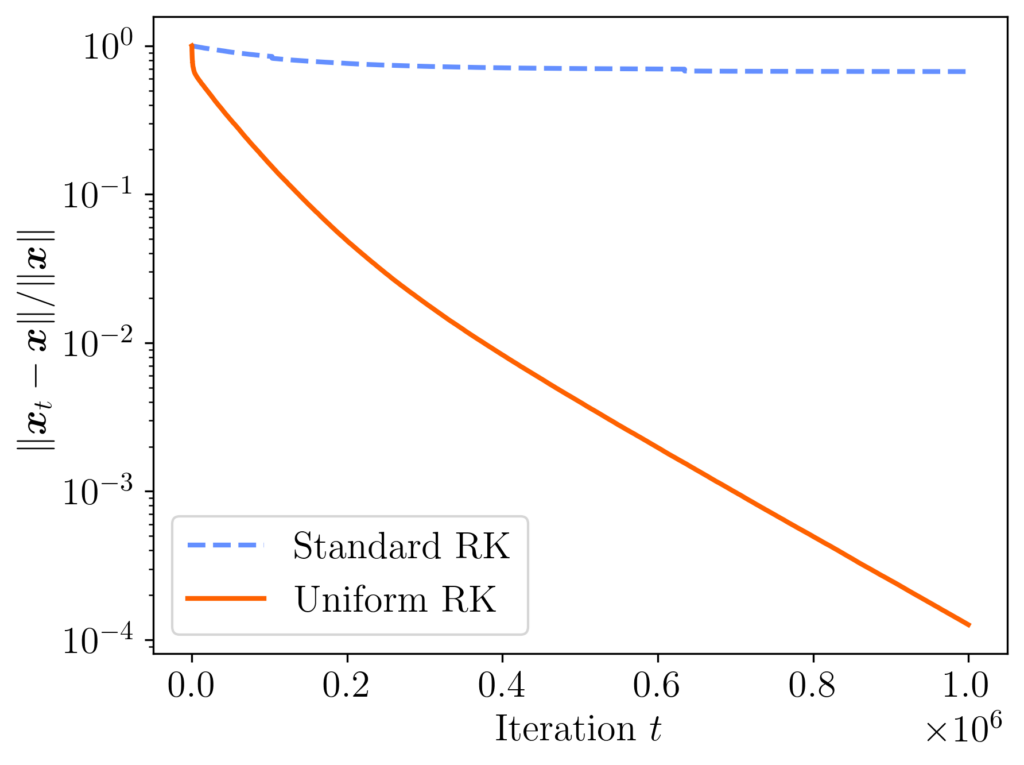
 , then the performance of both methods is similar, with both methods ending with a relative error of about 0.07.
, then the performance of both methods is similar, with both methods ending with a relative error of about 0.07.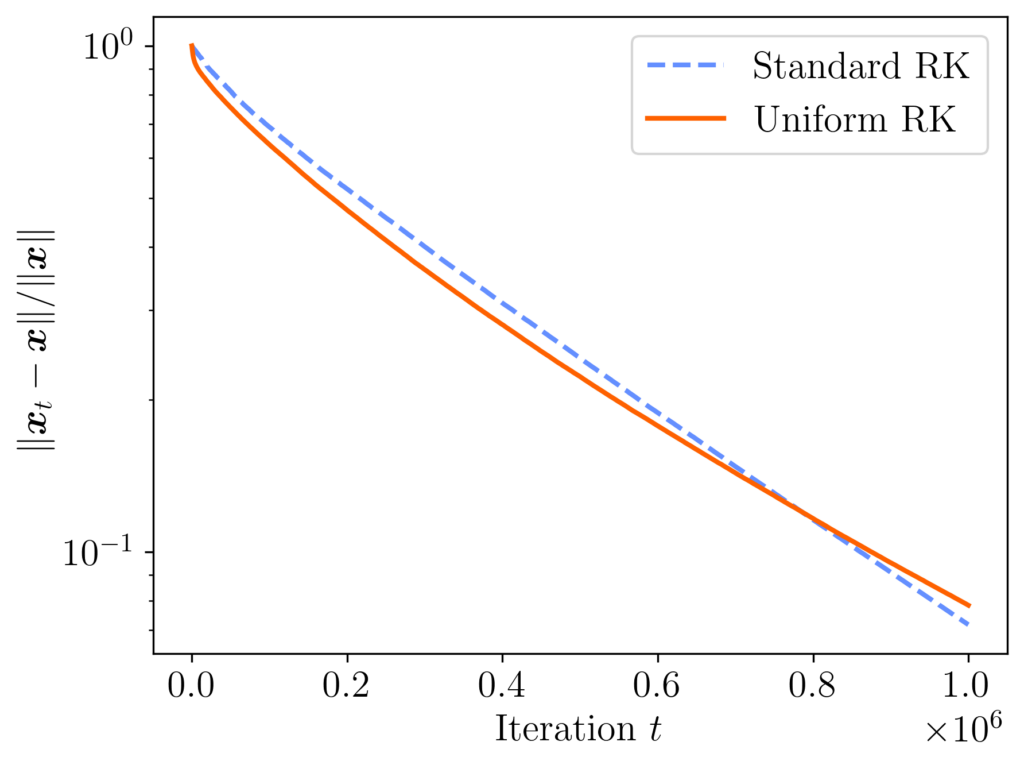
![\[\expect\left[ \norm{x_t - x_\star}^2 \right] \le (1 - \kappa_{\rm dem}(A)^{-2})^t \norm{x_\star}^2. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1f788737bb459ed2beaab020bca517c2_l3.png "Rendered by QuickLaTeX.com")
![\[\kappa_{\rm dem}(A) = \frac{\norm{A}_{\rm F}}{\sigma_{\rm min}(A)} = \sqrt{\sum_i \left(\frac{\sigma_i(A)}{\sigma_{\rm min}(A)}\right)^2} \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-824557fbf90de5be0f9756f9089728aa_l3.png "Rendered by QuickLaTeX.com")
 are the
are the  is consistent, possessing a solution
is consistent, possessing a solution  . If there are multiple solutions, we let
. If there are multiple solutions, we let  denote a diagonal matrix containing the inverse row norms, and introduce the row-equilibrated matrix
denote a diagonal matrix containing the inverse row norms, and introduce the row-equilibrated matrix  . The row-equilibrated matrix
. The row-equilibrated matrix  as standard RK applied to the row-equilibrated system
as standard RK applied to the row-equilibrated system  .
.![\[\expect\left[ \norm{\hat{x}_t - x_\star}^2 \right] \le (1 - \kappa_{\rm dem}(D_A A)^{-2})^t \norm{x_\star}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3fc6402842af70a33954d0133066a5e3_l3.png "Rendered by QuickLaTeX.com")
![\[\kappa(A) \coloneqq \frac{\sigma_{\rm max}(A)}{\sigma_{\rm min}(A)}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3fb2bba4d99c34bc055f7043fccfe19c_l3.png "Rendered by QuickLaTeX.com")
 . Also, let
. Also, let  denote the set of all (nonsingular) diagonal matrices.
denote the set of all (nonsingular) diagonal matrices.
 be wide
be wide  and full-rank, and let
and full-rank, and let  denote the row-scaling of
denote the row-scaling of ![\[\kappa_{\rm dem}(D_AA) \le \sqrt{n}\cdot \min_{D \in \mathrm{Diag}} \kappa (DA) \le \sqrt{n}\cdot \min_{D \in \mathrm{Diag}} \kappa_{\rm dem} (DA).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ec117b2c8c81cbcfc88eeae87eac0aaa_l3.png "Rendered by QuickLaTeX.com")
 factor of the optimal row scaling. In fact, we even bring the Demmel condition number to within a
factor of the optimal row scaling. In fact, we even bring the Demmel condition number to within a  , this result shows that implementing randomized Kaczmarz with uniform sampling yields to a convergence rate that is within a factor of
, this result shows that implementing randomized Kaczmarz with uniform sampling yields to a convergence rate that is within a factor of  denote the maximum ratio between two row norms:
denote the maximum ratio between two row norms: ![\[\gamma \coloneqq \frac{ \max_i \norm{a_i}}{\min_i \norm{a_i}}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9289f9e9a0f729efb516fbda78ac3453_l3.png "Rendered by QuickLaTeX.com")
![\[\kappa_{\rm dem}(A) \le \gamma \cdot \kappa_{\rm dem}(D_A A).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d842439abf3ace1c3c590d983ec36107_l3.png "Rendered by QuickLaTeX.com")
 , there exists a matrix
, there exists a matrix  where this bound is nearly attained:
where this bound is nearly attained: ![\[\kappa_{\rm dem}(A_\gamma) \ge \sqrt{1-\frac{1}{n}} \cdot \gamma \cdot \kappa_{\rm dem}(D_{A_\gamma}A_\gamma).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9ff68e369e8f22f56563fdc443894ce4_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{D_AA}_{\rm F} = \sqrt{n}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-dc083ac4d61189b0851e1495ff99f25d_l3.png "Rendered by QuickLaTeX.com")
 as follows
as follows ![\[\frac{1}{\sigma_{\rm min}(D_A A)} = \norm{A^\dagger D_A^{-1}}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c96cc4653bb180133ed40dd1c84687fd_l3.png "Rendered by QuickLaTeX.com")
 denotes the
denotes the  , we have
, we have ![\[\frac{1}{\sigma_{\rm min}(D_A A)} = \norm{A^\dagger D^{-1} (DD_A^{-1})} \le \norm{A^\dagger D^{-1}} \norm{DD_A^{-1}} = \frac{\norm{DD_A^{-1}}}{\sigma_{\rm min}(DA)}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a65986b5ef3ac5975280bffcd2fcb927_l3.png "Rendered by QuickLaTeX.com")
 is diagonal its spectral norm is
is diagonal its spectral norm is ![\[\norm{DD_A^{-1}} = \max \left\{ \frac{|D_{ii}|}{|(D_A)_{ii}|} : 1\le i \le n \right\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-705ca33a5372c0bdf858770f9d204436_l3.png "Rendered by QuickLaTeX.com")
 are
are  , so
, so ![\[\norm{DD_A^{-1}} = \max \left\{ |D_{ii}|\norm{a_i} : 1\le i\le n \right\}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b8026880c5418d6aac0894b839079753_l3.png "Rendered by QuickLaTeX.com")
 . The maximum row norm is always less than the largest singular value of
. The maximum row norm is always less than the largest singular value of  . Therefore, combining this result, (7), and (9), we obtain
. Therefore, combining this result, (7), and (9), we obtain ![\[\kappa_{\rm dem}(D_AA) \le \sqrt{n} \cdot \frac{\sigma_{\rm max}(DA)}{\sigma_{\rm min}(DA)} = \sqrt{n}\cdot \kappa (DA).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9e73680b6f4fe21817d43adb57a457ff_l3.png "Rendered by QuickLaTeX.com")
 , we are free to minimize over
, we are free to minimize over ![\[\kappa_{\rm dem}(D_AA) \le \sqrt{n}\cdot \min_{D \in \mathrm{Diag}} \kappa (DA).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e18d74284219efc70ced6833db98a733_l3.png "Rendered by QuickLaTeX.com")
 . Using the Moore–Penrose pseudoinverse again, write
. Using the Moore–Penrose pseudoinverse again, write ![\[\kappa_{\rm dem}(A) = \norm{D_A^{-1}(D_AA)}_{\rm F} \norm{(D_A A)^\dagger D_A}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-50cf827241cc20f905230e4a51946d0b_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{BC}_{\rm F} \le \norm{B}\norm{C}_{\rm F}, \quad \norm{BC} \le\norm{B}\norm{C}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-051f8f43c351dcc02a4d3342d55b5455_l3.png "Rendered by QuickLaTeX.com")
![\[\kappa_{\rm dem}(A) \le \norm{D_A^{-1}}\norm{D_AA}_{\rm F} \norm{(D_A A)^\dagger} \norm{D_A}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1d3aa06275558feecad7fe4a38c3f64a_l3.png "Rendered by QuickLaTeX.com")
 and
and  . We conclude
. We conclude  . Then
. Then  with
with  and
and ![\[\kappa_{\rm dem}(A_\gamma) = \frac{\norm{A_{\gamma}}_{\rm F}}{\sigma_{\rm min}(A_\gamma)} = \frac{\sqrt{(n-1)\gamma^2+1}}{1} \ge \sqrt{n} \cdot \sqrt{1-\frac{1}{n}} \cdot \gamma.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0216b29d23ab394e7040423bd9a13ebe_l3.png "Rendered by QuickLaTeX.com")
 more iterations than standard RK on a worst-case example, which can be a big difference for large problems. But, particularly for badly row-scaled problems, Proposition 2 shows that uniform RK can dramatically outcompete standard RK. Ultimately, I would give two answers.
more iterations than standard RK on a worst-case example, which can be a big difference for large problems. But, particularly for badly row-scaled problems, Proposition 2 shows that uniform RK can dramatically outcompete standard RK. Ultimately, I would give two answers. of two
of two  is positive semidefinite (psd, for short) if it is
is positive semidefinite (psd, for short) if it is  for all vectors
for all vectors  . All matrices in this post are real, though the proofs we’ll consider also extend to complex matrices. The entrywise product will be denoted
. All matrices in this post are real, though the proofs we’ll consider also extend to complex matrices. The entrywise product will be denoted  and is defined as
and is defined as  . The entrywise product is also known as the Hadamard product or Schur product.
. The entrywise product is also known as the Hadamard product or Schur product. :
: ![\[x^\top (A\circ M)x = \sum_{i,j=1}^n x_i (A\circ M)_{ij} x_j = \sum_{i,j=1}^n x_i A_{ij} M_{ij} x_j.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ad02e17a6c90aca9b469ef0fe73bb1d3_l3.png "Rendered by QuickLaTeX.com")
![\[x^\top (A\circ M)x = \sum_{i,j=1}^n x_i A_{ij} x_j M_{ji} = \tr(\operatorname{diag}(x) A \operatorname{diag}(x) M).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4386c5381ce8bcc4e8ded09b2c00bcf5_l3.png "Rendered by QuickLaTeX.com")
 ). Thus, we may write
). Thus, we may write  . Substituting these expressions in the trace formula and invoking the
. Substituting these expressions in the trace formula and invoking the ![\[x^\top (A\circ M)x = \tr(\operatorname{diag}(x) B^\top B \operatorname{diag}(x) C^\top C) = \tr(C\operatorname{diag}(x) B^\top B \operatorname{diag}(x) C^\top).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5f330914f0a1aa93654b4330e2615c7b_l3.png "Rendered by QuickLaTeX.com")
![\[C\operatorname{diag}(x) B^\top B \operatorname{diag}(x) C^\top = G^\top G \quad \text{for } G = B \operatorname{diag}(x) C^\top.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-27588c5bd3897e90305feba5e03a6488_l3.png "Rendered by QuickLaTeX.com")
![\[x^\top (A\circ M)x = \tr(G^\top G) \ge 0.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c7e68b9a2fc38f723e37e208c2d4c28b_l3.png "Rendered by QuickLaTeX.com")
 for every vector
for every vector  and
and  denote the
denote the  and
and  , we have
, we have ![\[A = \sum_i b_ib_i^\top \quad \text{and} \quad M = \sum_j c_jc_j^\top.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-95d0f789d3f2d56faa06e53e7bc33366_l3.png "Rendered by QuickLaTeX.com")
![\[A\circ M = \sum_{i,j} (b_ib_i^\top \circ c_jc_j^\top).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c808a8bf106990528aae134391107813_l3.png "Rendered by QuickLaTeX.com")
 and
and  is, by direct computation,
is, by direct computation,  . Thus,
. Thus, ![\[A\circ M = \sum_{i,j} (b_i\circ c_j)(b_i\circ c_j)^\top\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5484fa9e5570cb1c2a639512b48c303c_l3.png "Rendered by QuickLaTeX.com")
 be
be  is seen to have zero mean as well. Thus, the
is seen to have zero mean as well. Thus, the  entry of the covariance matrix
entry of the covariance matrix  of
of ![\[\expect[x_iy_ix_jy_j] = \expect[x_ix_j] \expect[y_iy_j] = A_{ij} M_{ij} = (A\circ M)_{ij}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b792b112f5388bf21a7f6254af9b271d_l3.png "Rendered by QuickLaTeX.com")
 have replaced by expectations
have replaced by expectations ![A = \expect [xx^\top]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ec158da95565c147435a98aff8fdd42c_l3.png "Rendered by QuickLaTeX.com") .
. of two psd matrices
of two psd matrices ![\[A\circ M = ((A\otimes M)_{(i+n(i-1))(i+n(i-1))} : i = 1,\ldots,n).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-46b3a14143b4f365e5cd92118733a1bb_l3.png "Rendered by QuickLaTeX.com")
 be vectors, assemble the matrix
be vectors, assemble the matrix  , and form the
, and form the ![\[A = B^\top B = \onebytwo{x}{y}^\top \onebytwo{x}{y} = \twobytwo{\norm{x}^2}{y^\top x}{x^\top y}{\norm{y}^2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4298058f076817cf981a6d027419d2d9_l3.png "Rendered by QuickLaTeX.com")
![\[\det(A) = \norm{x}^2\norm{y}^2 - |x^\top y|^2 \ge 0.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4395b6aa06933ae0e57f5b542742c2e9_l3.png "Rendered by QuickLaTeX.com")
![\[|x^\top y| \le \norm{x}\norm{y}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2d87e0f0225bbe13645f83b262e48ee4_l3.png "Rendered by QuickLaTeX.com")
 be strictly positive numbers. Then the inverse of their average is no bigger than the average of their inverses:
be strictly positive numbers. Then the inverse of their average is no bigger than the average of their inverses: ![\[\left( \frac{1}{n} \sum_{i=1}^n x_i \right)^{-1} \le \frac{1}{n} \sum_{i=1}^n \frac{1}{x_i}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-db85840bf998e96281f1c2605ca740c5_l3.png "Rendered by QuickLaTeX.com")
 into a
into a  positive semidefinite matrix
positive semidefinite matrix  . Taking the average of all such matrices, we observe that
. Taking the average of all such matrices, we observe that ![\[A = \twobytwo{\frac{1}{n} \sum_{i=1}^n x_i}{1}{1}{\frac{1}{n} \sum_{i=1}^n \frac{1}{x_i}}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8b30ffa6031c16bca1ca7be4669236ed_l3.png "Rendered by QuickLaTeX.com")
![\[\det(A) = \left(\frac{1}{n} \sum_{i=1}^n x_i\right) \left(\frac{1}{n} \sum_{i=1}^n \frac{1}{x_i}\right) - 1 \ge 0.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e1e6cd521eaf4f49c17bab509cf71cd2_l3.png "Rendered by QuickLaTeX.com")
 is a matrix and
is a matrix and  is a vector. For simplicity, we assume that this system is
is a vector. For simplicity, we assume that this system is  ). The
). The  , randomized Kaczmarz repeatedly performs the following steps:
, randomized Kaczmarz repeatedly performs the following steps: according to the probability distribution
according to the probability distribution  . Here, and going forward,
. Here, and going forward,  denotes the
denotes the  is the
is the  .
. are not very large, this is an easy problem: Simply sweep through all the rows
are not very large, this is an easy problem: Simply sweep through all the rows  and compute their norms
and compute their norms  . Then, sampling can be done using any algorithm for sampling from a weighted list of items. But what if
. Then, sampling can be done using any algorithm for sampling from a weighted list of items. But what if ![\[\norm{a_i}^2 \le B \quad \text{for each } i = 1,\ldots,n.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a80c9da38c2d98b188cb70f64c8637e8_l3.png "Rendered by QuickLaTeX.com")
 . To sample a row
. To sample a row  , we perform the following the rejection sampling procedure:
, we perform the following the rejection sampling procedure:
 uniformly at random (i.e.,
uniformly at random (i.e.,  is equally likely to be any row index between
is equally likely to be any row index between  , accept and set
, accept and set  . With the remaining probability
. With the remaining probability  , reject and return to step 1.
, reject and return to step 1. . Our goal is to choose a random index
. Our goal is to choose a random index  , i.e.,
, i.e.,  . We will call
. We will call  the target distribution. (Note that we do not require the weights
the target distribution. (Note that we do not require the weights  .)
.) , i.e., we can efficiently generate random
, i.e., we can efficiently generate random  . Further, suppose that the proposal distribution dominates the target distribution in the sense that
. Further, suppose that the proposal distribution dominates the target distribution in the sense that ![\[w_i \le \rho_i \quad \text{for each } i=1,\ldots,n.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7f2901cd8e565853465062e41a59b5bd_l3.png "Rendered by QuickLaTeX.com")
 , accept and set
, accept and set  and
and  for all
for all  , after which the probability of accepting
, after which the probability of accepting  . Therefore, the probability of outputting
. Therefore, the probability of outputting ![\[\prob \{\text{$i$ accepted this loop}\} = \frac{\rho_i}{\sum_{j=1}^n \rho_j} \cdot \frac{w_i}{\rho_i} = \frac{w_i}{\sum_{j=1}^n \rho_j}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f8fb5712ddc38527600d4fd51394bfc7_l3.png "Rendered by QuickLaTeX.com")
![\[\prob \{\text{any accepted this loop}\} = \sum_{i=1}^n \prob \{\text{$i$ accepted this loop}\} = \frac{\sum_{i=1}^n w_i}{\sum_{i=1}^n \rho_i}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-87004ae4b99269f6917de03d3a4ec86e_l3.png "Rendered by QuickLaTeX.com")
![\[\prob \{\text{$i$ accepted this loop} \mid \text{any accepted this loop}\} = \frac{\prob \{\text{$i$ accepted this loop}\}}{\prob \{\text{any accepted this loop}\}} = \frac{w_i}{\sum_{j=1}^n w_j}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8740003cf2cc5dfd18def3387663bcc2_l3.png "Rendered by QuickLaTeX.com")
 , the ratio of the total mass of the target
, the ratio of the total mass of the target  . Thus, rejection sampling will have a high acceptance rate if
. Thus, rejection sampling will have a high acceptance rate if  and a low acceptance rate if
and a low acceptance rate if  . The conclusion for practice is that rejection sampling is only computationally efficient if one has access to a good proposal distribution
. The conclusion for practice is that rejection sampling is only computationally efficient if one has access to a good proposal distribution  .
. , but this not all that rejection sampling can do. Indeed, one can also use rejection sampling for sampling a real-valued parameters
, but this not all that rejection sampling can do. Indeed, one can also use rejection sampling for sampling a real-valued parameters  or a multivariate parameter
or a multivariate parameter  from a given (unnormalized) probability density function (or, if one likes, an unnormalized probability measure).
from a given (unnormalized) probability density function (or, if one likes, an unnormalized probability measure). and
and  are necessary to define the sampling probabilities
are necessary to define the sampling probabilities  and
and  , as computing
, as computing  requires a full pass over the data to compute.
requires a full pass over the data to compute. denote the initial matrix and
denote the initial matrix and  denote a trivial initial approximation. For
denote a trivial initial approximation. For  , RPCholesky performs the following steps:
, RPCholesky performs the following steps: with probability
with probability  .
. . Here,
. Here,  denotes the
denotes the  .
. .
. . With an optimized implementation, RPCholesky requires only
. With an optimized implementation, RPCholesky requires only  operations and evaluates just
operations and evaluates just  entries of the input matrix
entries of the input matrix 
 denote the first
denote the first  produced by
produced by ![\[\hat{A}^{(j)} = A(:,S_j) A(S_j,S_j)^{-1}A(S_j,:). \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ebc63948bd7d20eda4ccafa74555ba6b_l3.png "Rendered by QuickLaTeX.com")
![\[A^{(j)} = A - \hat{A}^{(j)} = A - A(:,S_j) A(S_j,S_j)^{-1}A(S_j,:).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-dc5136dd307c099cb5fa70663104166b_l3.png "Rendered by QuickLaTeX.com")
![\[A^{(j)}_{ii} = A_{ii} - A(i,S_j) A(S_j,S_j)^{-1} A(S_j,i).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f8442521e6a6003a935f35d25aca1c73_l3.png "Rendered by QuickLaTeX.com")
 is cheap, just requiring some arithmetic involving matrices and vectors of size
is cheap, just requiring some arithmetic involving matrices and vectors of size  , but it is expensive to evaluate all of the
, but it is expensive to evaluate all of the  . (One may verify that, as required,
. (One may verify that, as required,  for all
for all  , sample random indices
, sample random indices  using rejection sampling with proposal distribution
using rejection sampling with proposal distribution  are evaluated on an as-needed basis using (2).
are evaluated on an as-needed basis using (2). and
and  . The first matrix
. The first matrix  to its
to its  . The other matrix
. The other matrix  . Let’s see how long it takes to compute their eigenvalue decompositions in MATLAB:
. Let’s see how long it takes to compute their eigenvalue decompositions in MATLAB: longer to compute the eigenvalues of the non-Hermitian matrix
longer to compute the eigenvalues of the non-Hermitian matrix  of eigenvectors for a Hermitian matrix
of eigenvectors for a Hermitian matrix  is a
is a  .
. . The matrix
. The matrix  of eigenvectors for a normal matrix
of eigenvectors for a normal matrix  is also unitary,
is also unitary,  . An important class of normal matrices are unitary matrices themselves. A unitary matrix
. An important class of normal matrices are unitary matrices themselves. A unitary matrix  is always normal since it satisfies
is always normal since it satisfies  .
.![\[C = H + iS, \quad H = \frac{C+C^*}{2}, \quad S = \frac{C-C^*}{2i}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5efdc333ce38cd9aaa9c5c10ad40b44a_l3.png "Rendered by QuickLaTeX.com")
 and
and  . Both
. Both  . We know from matrix theory that commuting Hermitian matrices are
. We know from matrix theory that commuting Hermitian matrices are  such that
such that  and
and  for diagonal matrices
for diagonal matrices  and
and  . Thus, given access to such
. Thus, given access to such ![\[C = Q(D_H+iD_S)Q^*.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d84fd3e79327e2e4082ba99967de3c9e_l3.png "Rendered by QuickLaTeX.com")
 for this matrix has a repeated eigenvalue. Thus,
for this matrix has a repeated eigenvalue. Thus,  of the Hermitian and skew-Hermitian parts of
of the Hermitian and skew-Hermitian parts of  and
and  .
. unitary matrix, RandDiag takes 0.4 seconds, just as fast as the Hermitian eigendecomposition did.
unitary matrix, RandDiag takes 0.4 seconds, just as fast as the Hermitian eigendecomposition did. , a matrix
, a matrix  will also diagonalize
will also diagonalize  . But, for any specific choice of
. But, for any specific choice of  , there is a possibility of failure. To avoid this possibility, we can just pick
, there is a possibility of failure. To avoid this possibility, we can just pick  and
and  at random. It’s really as simple as that.
at random. It’s really as simple as that. with
with  , its (
, its ( , where
, where  is a
is a  is
is  test matrix. It takes about 2.5 seconds to run.
test matrix. It takes about 2.5 seconds to run. is the
is the  is also small:
is also small: , computing its (upper triangular)
, computing its (upper triangular)  , and setting
, and setting  . Cholesky QR is very fast, about
. Cholesky QR is very fast, about  faster than Householder QR for this example:
faster than Householder QR for this example: , about ten million times larger than for Householder QR!:
, about ten million times larger than for Householder QR!: . The condition number of
. The condition number of  , which is at the root of Cholesky QR’s loss of accuracy. Thus, Cholesky QR is only appropriate for matrices that are well-conditioned, having a small condition number
, which is at the root of Cholesky QR’s loss of accuracy. Thus, Cholesky QR is only appropriate for matrices that are well-conditioned, having a small condition number  , say
, say  .
. that
that  is well-conditioned. Then, since
is well-conditioned. Then, since  ; see
; see  . This step compresses the very tall matrix
. This step compresses the very tall matrix  to the much shorter matrix
to the much shorter matrix  .
. using Householder QR. Since the matrix
using Householder QR. Since the matrix  .
. . Observe that
. Observe that  , as desired.
, as desired. faster in our experiment:
faster in our experiment: of an
of an  . The classical method for this purpose is the
. The classical method for this purpose is the ![\[\hat{\tr} = \frac{1}{m} \left( x_1^\top Ax_1 + \cdots + x_m^\top Ax_m \right),\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f27a0c05da428b96172a0a8ab23ca54b_l3.png "Rendered by QuickLaTeX.com")
 are
are ![\[\expect[x_ix_i^\top] = I.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b5ac53011350edc256d9b8c66fdbe7c8_l3.png "Rendered by QuickLaTeX.com")
 with equal probabilities (i.e.,
with equal probabilities (i.e.,  .
.![\expect[\hat{\tr}] = \tr(A)](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ad9e9d417b59555f2a6e78e78b6bcbe9_l3.png "Rendered by QuickLaTeX.com") , the
, the  is equal to the
is equal to the  of the Girard–Hutchinson estimator with different choices of test vectors. In that post, I stated the formulas for different choices of test vectors (Gaussian, random signs, sphere) and showed how those formulas could be proven.
of the Girard–Hutchinson estimator with different choices of test vectors. In that post, I stated the formulas for different choices of test vectors (Gaussian, random signs, sphere) and showed how those formulas could be proven. be the
be the ![\[\overline{\lambda} = \frac{\lambda_1 + \cdots + \lambda_n}{n}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3f9e9cf25f6cf20463de5be7e8d40f88_l3.png "Rendered by QuickLaTeX.com")
![\[\Var(\hat{\tr}_{\rm Gaussian}) = \frac{1}{m} \cdot 2 \sum_{i=1}^n \lambda_i^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f8dc441dc68294b0d6ff6af42d3ecef5_l3.png "Rendered by QuickLaTeX.com")
![\[\Var(\hat{\tr}_{\rm sphere}) = \frac{1}{m} \cdot \frac{n}{n+2} \cdot 2\sum_{i=1}^n (\lambda_i - \overline{\lambda})^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6a275f8ba8a47be085e22f679b60b497_l3.png "Rendered by QuickLaTeX.com")
 is smaller than
is smaller than  by a factor of
by a factor of  . This improvement is quite minor. Second, and more importantly,
. This improvement is quite minor. Second, and more importantly,  with a (
with a ( , we take
, we take  . Below show the variance of Girard–Hutchinson estimator for different distributions for the test vector. We see that the sphere distribution leads to a trace estimate which has a variance 300× smaller than the Gaussian distribution. For this example, the sphere and random sign distributions are similar.
. Below show the variance of Girard–Hutchinson estimator for different distributions for the test vector. We see that the sphere distribution leads to a trace estimate which has a variance 300× smaller than the Gaussian distribution. For this example, the sphere and random sign distributions are similar. )
)

![\[\Var(\hat{\tr}_{\rm signs}) = 2 \sum_{i\ne j} A_{ij}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3a6665f8e55563640aedd5ebb0945409_l3.png "Rendered by QuickLaTeX.com")
 depends on the size of the off-diagonal entries of
depends on the size of the off-diagonal entries of