Today, I want to talk about the generalized Nyström approximation, which I regard as the one of the “big three” approaches to constructing a low-rank approximation to matrix.1The other two approaches are the “projection approximation”/randomized SVD approach and the Nyström approximation. Understanding this approximation, under what conditions it works and the sharpest possible error bounds for it, is a subject of two recent papers of mine:
- Faster Randomized Linear Algebra with Structured Random Matrices, joint with Chris Camaño, Raphael Meyer, and Joel Tropp.
- Sharp analysis of sketched least squares and randomized low-rank approximation, joint with Robert Webber.
On the occasion of the release of the second paper this morning, I felt it was a good time to talk about the generalized Nyström approximation on this blog. In this post, I will try and motivate the generalized Nyström approximation, describing the motivation for the method and when it might be preferable to alternatives.
Existing Characters: Nyström Approximation and the Randomized SVD
To begin our story, let me begin with a reminder of a couple of characters we’ve met in previous installments of this blog, randomized Nyström approximation and the randomized SVD.
Randomized Nyström approximation is a method for producing a low-rank approximation to a positive semidefinite2For this post, a positive semidefinite matrix will always be real symmetric or complex Hermitian. We will focus only on real matrices for this post, though the extension to complex matrices is straightforward. (psd) matrix  . For form this approximation, begin by drawing a random test matrix
. For form this approximation, begin by drawing a random test matrix  , say, with independent standard normal random entries. (We will have more to say about the choice of below). Using this test matrix, the Nyström approximation is defined as3Here, the inverse
, say, with independent standard normal random entries. (We will have more to say about the choice of below). Using this test matrix, the Nyström approximation is defined as3Here, the inverse  should be interpreted as a Moore–Penrose pseudoinverse in the event where
should be interpreted as a Moore–Penrose pseudoinverse in the event where  is singular.
is singular.
![\[\hat{A} = A\Omega (\Omega^\top A\Omega)^{-1} (A\Omega)^\top.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-858ee4e7300bc0e48d12677bd459aeaf_l3.png "Rendered by QuickLaTeX.com")
The randomized SVD is a method for constructing a low-rank approximation to a general, non-symmetric or even non-square matrix  . Again, begin by constructing a random test matrix . To construct a low-rank approximation, compute the product
. Again, begin by constructing a random test matrix . To construct a low-rank approximation, compute the product  and orthonormalize its columns (e.g., by QR decomposition) to obtain
and orthonormalize its columns (e.g., by QR decomposition) to obtain
![\[Q \coloneqq \operatorname{orth}(B\Omega).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-77d0237ae3f34369630eff1996a39563_l3.png "Rendered by QuickLaTeX.com")
, yielding the low-rank approximation ![\[\hat{B} = QC \quad \text{for } C = Q^\top B.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f9906f14f437fa982d3c8a2311d36ee3_l3.png "Rendered by QuickLaTeX.com")
How do these two algorithms compare? There are at least three major differences between the two algorithms. Here are the first two:
- Scope. Nyström approximation applies only to psd matrices, and randomized SVD applies to a general rectangular mtrix.
- Single-pass? The Nyström approximation requires only a single pass over the matrix to form. (Each entry of needs to be read once to form the product
 , after which we have all the information we need from to form
, after which we have all the information we need from to form  .) By contrast, the randomized SVD requires two passes, one to compute and a second to compute
.) By contrast, the randomized SVD requires two passes, one to compute and a second to compute  .
.
The third point is more subtle and concerns the accuracy of these algorithms. As we saw in a previous post, the randomized SVD approximation satisfies the error bound
(1) ![\[\expect \norm{B - \hat{B}}_{\rm F}^2 \le \min_{r \le k-2} \left( 1 + \frac{r}{k-(r+1)} \right) \norm{B - \lowrank{B}_r}_{\rm F}^2. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e3bfd87c8b50495a7a32c42f0af681ac_l3.png "Rendered by QuickLaTeX.com")
 is the matrix Frobenius norm and
is the matrix Frobenius norm and  denotes the best rank-
denotes the best rank- approximation to . This result, due to Halko, Martinsson, & Tropp (2011), shows that the error of rank-
approximation to . This result, due to Halko, Martinsson, & Tropp (2011), shows that the error of rank- randomized SVD is comparable to the error of the best rank- approximation to of any rank
randomized SVD is comparable to the error of the best rank- approximation to of any rank  . See this post for more discussion of this error bound.
. See this post for more discussion of this error bound.
Here is analogous bound for the randomized Nyström approximation, taken from Corollary 8.3 in this paper of Tropp and Webber:
(2) ![\[\left(\expect \norm{A - \hat{A}}_{\rm F}^2 \right)^{1/2} \le \min_{r\le k-4} \left(1 + \frac{r+1}{k-(r+3)}\right) \left( \norm{A - \lowrank{A}}_{\rm F} + \frac{1}{\sqrt{k-r}} \norm{A - \lowrank{A}_r}_* \right). \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-af14e93166cedc601f5dee4cc4155a7a_l3.png "Rendered by QuickLaTeX.com")
 of the best rank- approximation.
of the best rank- approximation.
The nuclear norm
![\[\norm{C}_* = \sigma_1(C) + \sigma_2(C) + \cdots\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-70e4f079cb458b88416ee8f1c2f9ec2b_l3.png "Rendered by QuickLaTeX.com")
 decrease a slow rate. The matrix with the slowest-possible rate of singular decrease is the identity matrix. For this matrix, its Frobenius is
decrease a slow rate. The matrix with the slowest-possible rate of singular decrease is the identity matrix. For this matrix, its Frobenius is  , and its nuclear norm is
, and its nuclear norm is  —a factor
—a factor  larger!
larger!
The conclusion of this discussion is that, for matrices with slowly decaying eigenvalues4Recall that the eigenvalues and singular values of a psd matrix coincide., the the randomized Nyström error bound (2) can be much larger than the randomized SVD error bound (1). For such problems, the error of the randomized SVD can be much smaller than the error of randomized Nyström approximation. We add this to our list of comparisons
- Frobenius-norm error bounds? The (Frobenius-norm) error of the randomized SVD
 is bounded in terms of the Frobenius-norm error of the best rank- approximation
is bounded in terms of the Frobenius-norm error of the best rank- approximation  for
for  . For the Nyström approximation, the error
. For the Nyström approximation, the error  is bounded by a more complicated expression that also involves the nuclear norm of the best rank- approximation.
is bounded by a more complicated expression that also involves the nuclear norm of the best rank- approximation.
Generalized Nyström Approximation: The Best of Both Worlds
It is natural to ask: Is there one algorithm that achieves the positive attributes of both the randomized Nyström and randomized SVD algorithms? Is there a single-pass low-rank approximation algorithm that can be applying to any rectangular matrix and achieves Frobenius-norm error bounds? The answer is yes, and we will derive such an approximation now.
As with the randomized SVD and randomized Nyström approximation, we first compute the product  of with a random matrix . We may then search for the best approximation
of with a random matrix . We may then search for the best approximation  to spanned by the columns of
to spanned by the columns of  . Such an approximation takes the form
. Such an approximation takes the form  , and we may find the best
, and we may find the best  by solving a least-squares problem
by solving a least-squares problem
(3) ![\[W = \argmin_W \norm{B - YW}_{\rm F}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c10c5948a3d56992543344b0490d9a1a_l3.png "Rendered by QuickLaTeX.com")
 , where
, where  is the Moore–Penrose pseudoinverse. The resulting low-rank approximation is
is the Moore–Penrose pseudoinverse. The resulting low-rank approximation is  . In fact, this approximation coincides with the approximation generated by the randomized SVD algorithm. As with the standard randomized SVD, this approximation takes two passes over to form, one to form and a second to form .
. In fact, this approximation coincides with the approximation generated by the randomized SVD algorithm. As with the standard randomized SVD, this approximation takes two passes over to form, one to form and a second to form .
To obtain a one-pass algorithm, we need a faster way of computing an (approximate) solution to the least-squares problem (3). As we have seen before on this blog, sketching provides a natural approach to quickly and approximately solving a least-squares problem. Specifically, we draw another random test matrix  and solve the “sketched” least squares problem
and solve the “sketched” least squares problem
(4) ![\[\hat{W} = \argmin_W \norm{\Phi^\top B - (\Phi^\top Y)W}_{\rm F}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e07e026f85c1bc718d95c5c88a47d036_l3.png "Rendered by QuickLaTeX.com")
 of the sketching matrix such be larger than the rank , e.g..
of the sketching matrix such be larger than the rank , e.g..  . The solution to (4) is
. The solution to (4) is ![\[\hat{W} = (\Phi^\top Y)^\dagger (\Phi^\top B)\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-bdf37e0344474a81247242dc1fbc106f_l3.png "Rendered by QuickLaTeX.com")
![\[\hat{B} = Y(\Phi^\top Y)^\dagger (\Phi^\top B) = (B\Omega) (\Phi^\top B\Omega)^\dagger (\Phi^\top B).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e2fdc92bd90711c249030489953513de_l3.png "Rendered by QuickLaTeX.com")
. (Namely, one should acquire—in the same pass—the products and  .) The generalized Nyström approximation also satisfies our other desired properties, applying to general, rectangular matrix and, as we will see, achieving Frobenius-norm approximation error.
.) The generalized Nyström approximation also satisfies our other desired properties, applying to general, rectangular matrix and, as we will see, achieving Frobenius-norm approximation error.
History
In the modern randomized linear algebra literature, the generalized Nyström approximation appears to have been concurrently discovered by Woolfe, Liberty, Rokhlin, & Tygert (2008) and Clarkson & Woodruff (2009). An algebraically equivalent but more numerically stable version of the generalized Nyström approximation was developed by Tropp, Yurtsever, Udell, & Cevher (2017). Nakatsukasa (2020) re-examined the low-rank approximation format, developed a different class of numerical stable implementations, and suggested the name generalized Nyström approximation. Alex Townsend and Per-Gunnar Martinsson trace the origins of this low-rank approximation format far earlier back to the works of Wedderburn (1934).
Implementation
This post is concerned with the generalized Nyström approximation as a type of low-rank approximation format. To use generalized Nyström approximation in practice, one must use an appropriate algorithm which computes the decomposition in a stable way.
Perhaps the simplest algorithm for computing a generalized Nyström approximation was studied by Nakatsukasa (2020). One begins by computing the matrices
![\[Y = B\Omega, \quad Z = B^\top \Omega, \quad C = \Phi^\top Y.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d95eee60a18e0b58b304b250ab7f3b0f_l3.png "Rendered by QuickLaTeX.com")
as a factored matrix, one takes a QR decomposition  and defines
and defines  and
and  . The generalized Nyström approximation has been computed in factored form:
. The generalized Nyström approximation has been computed in factored form:  . In cases where the core matrix is rank-deficient up to machine precision, the numerical stability of this procedure can sometimes be aided by using a truncated SVD or column-pivoted QR decomposition of ; see Nakatsukasa’s paper for details. An alternate implementation which outputs as a compact SVD was developed by Tropp, Yurtsever, Udell, and Cevher (2017).
. In cases where the core matrix is rank-deficient up to machine precision, the numerical stability of this procedure can sometimes be aided by using a truncated SVD or column-pivoted QR decomposition of ; see Nakatsukasa’s paper for details. An alternate implementation which outputs as a compact SVD was developed by Tropp, Yurtsever, Udell, and Cevher (2017).
Relationship to Other Formats
As the name suggests, the generalized Nyström approximation format generalizes the Nyström approximation beyond psd matrices. Indeed, the standard Nyström approximation
![\[\hat{A} = (A\Omega) (\Omega^\top A \Omega)^\dagger (\Omega^\top A)\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-fb7ae8960f8c0173063c23f7290a1b63_l3.png "Rendered by QuickLaTeX.com")
with a test matrix  .
.
Perhaps less obviously, the generalized Nyström approximation also generalizes the randomized SVD approximation. Indeed, the randomized SVD approximation  is the generalized Nyström approximation with trivial right test matrix
is the generalized Nyström approximation with trivial right test matrix  .
.
Generalized Nyström Approximation = Sketch-and-Solve + Randomized SVD
What is the generalized Nyström approximation? There are several interpretations. For instance, if  and
and  is invertible, the generalized Nyström approximation is the unique approximation satisfying the interpolatory condition
is invertible, the generalized Nyström approximation is the unique approximation satisfying the interpolatory condition
![\[\hat{B}\Omega = B\Omega \quad \text{and} \quad \hat{B}^\top\Phi = B^\top\Phi.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ed39d951ca827f023b0460fce18b1e0a_l3.png "Rendered by QuickLaTeX.com")
Notwithstanding the validity and usefulness of other interpretations, my view is that the most useful interpretation of generalized Nyström approximation is the one we started with:
Generalized Nyström approximation is a sketched version of the randomized SVD approximation.
To see this insight in action, we will use it to analyze the generalized Nyström approximation with Gaussian test matrices. Let  and be populated with independent standard Gaussian random entries, and as we have been, assume
and be populated with independent standard Gaussian random entries, and as we have been, assume  . Let us analyze the expected (squared) Frobenius-norm error of the generalized Nyström approximation.
. Let us analyze the expected (squared) Frobenius-norm error of the generalized Nyström approximation.
We will use the following result for sketching with a Gaussian embedding, due to Bartan & Pilanci (2020) and discussed in this previous blog post.
Theorem 1 (sketch-and-solve): Consider a (matrix) least-squares problem
with dimensions
and
. Let
Then
![\[W = Y^\dagger B= \argmin_W \norm{B - YW}_{\rm F}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1fbec03b765e920f2546d70acc1e315d_l3.png "Rendered by QuickLaTeX.com")
![\[\hat{W} = (\Phi^\top Y)^\dagger \Phi^\top B = \argmin_W \norm{\Phi^\top B - (\Phi^\top Y)W}_{\rm F}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-47453e73c76d516332da7709bec4fdf2_l3.png "Rendered by QuickLaTeX.com")
![\[\expect \norm{B - Y\hat{W}}_{\rm F}^2 = \expect \norm{B - Y(\Phi^\top Y)^\dagger\Phi^\top B}_{\rm F}^2 = \left( 1 + \frac{k}{p - (k+1)} \right)\norm{B - YY^\dagger B}_{\rm F}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1dd58766c5711cb52838468de28f2956_l3.png "Rendered by QuickLaTeX.com")
We can apply this result to the generalized Nyström approximation by setting . Let  denote the expectation over
denote the expectation over  alone. Then
alone. Then
![\[\expect_\Phi \norm{B - B\Omega(\Phi^\top B\Omega)^\dagger\Phi^\top B}_{\rm F}^2 = \left( 1 + \frac{k}{p - (k + 1)} \right)\norm{B - (B\Omega)(B\Omega)^\dagger B}_{\rm F}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3ca90719321990c2e97c775f86238ce7_l3.png "Rendered by QuickLaTeX.com")
 is just the randomized SVD approximation. Invoking the randomized SVD bound (1) yields
is just the randomized SVD approximation. Invoking the randomized SVD bound (1) yields ![\begin{align*}\expect \norm{B - B\Omega(\Phi^\top B\Omega)^\dagger\Phi^\top B}_{\rm F}^2 &= \expect_\Omega\left[\expect_\Phi \norm{B - B\Omega(\Phi^\top B\Omega)^\dagger\Phi^\top B}_{\rm F}^2\right] \\&= \left( 1 + \frac{k}{p - (k + 1)} \right)\expect_\Omega \norm{B - (B\Omega)(B\Omega)^\dagger B}_{\rm F}^2 \\&\le \left( 1 + \frac{k}{p - (k + 1)} \right)\left[\min_{r < k-1}\left( 1 + \frac{r}{k - (r + 1)} \right) \norm{B - \lowrank{B}_r}_{\rm F}^2\right].\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f6a0cfb074737e67fcb3b172f5d5badc_l3.png "Rendered by QuickLaTeX.com")
Theorem 2 (generalized Nyström approximation): With the present setting, it holds that
![\[\expect \norm{B - B\Omega(\Phi^\top B\Omega)^\dagger\Phi^\top B}_{\rm F}^2 \le \left( 1 + \frac{k}{p - (k + 1)} \right)\left[\min_{r < k-1}\left( 1 + \frac{r}{k - (r + 1)} \right) \norm{B - \lowrank{B}_r}_{\rm F}^2\right].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-be78edd2761af9a1cc467056a24c73e9_l3.png "Rendered by QuickLaTeX.com")
This result is Theorem 4.3 in this paper of Tropp, Yurtsever, Udell, & Cevher (2017). A slight refinement of this bound appears in my paper with Robert Webber, and we show that our new bound is sharp on hard examples. Thus, Theorem 2 is nearly the best possible error bound for the generalized Nyström approximation.
Choice of Random Matrix
For most of this post, we have focused on the cases where the random test matrices and are unstructured matrices with Gaussian random entries. But can we use more structured random test matrices? Say, sparse test matrices? Do these lead to faster low-rank approximation algorithms?
For the randomized SVD, the results are disappointing. Computing with a sparse random matrix is fast. But then we compute  and compute
and compute  ; the matrix
; the matrix  is dense and unstructured, so computing is slower and all benefits of the sparse test matrix have been erased.
is dense and unstructured, so computing is slower and all benefits of the sparse test matrix have been erased.
The issue with the randomized SVD is that it’s a two-pass algorithm: The first pass, computing , can be done using a sparse random test matrix. But the second pass requires a matrix product with a dense matrix .
The situation is much better for the generalized Nyström approximation, which requires only a single pass and can be implemented only by multiplying against sparse matrices. Indeed, generating and to be sparse matrices, the generalized Nyström approximation can be written
![\[\hat{B} = Y(\Phi^* Y)^\dagger Z \quad \text{for } Y = B\Omega \text{ and } Z = \Phi^\top B.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-959c9e287d25f46bd1011f38affcab7b_l3.png "Rendered by QuickLaTeX.com")
has been isolated into matrix products with the random test matrices, and we obtain speedups by replacing using sparse random test matrices for and .
Structured test matrices, like sparse ones, can be very powerful. But basic theoretical questions remain about their properties. We tackle these theoretical questions in my new paper (joint with Chris Camaño, Raphael Meyer, and Joel Tropp), and we provide experiments demonstrating how structured sketching matrices can lead to large speedups in generalized Nyström approximation and other linear algebra tasks. I think it’s a really neat paper, and my wonderful collaborator Chris did some really beautiful experiments for it. I hope you’ll check it out!
 be random vectors and let
be random vectors and let  denote their
denote their  are
are ![\[\expect[x_i^{\vphantom{\top}} x_i^\top] = I.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5e65b0d7cf66e1cecc4c60470eca40af_l3.png "Rendered by QuickLaTeX.com")
 inherits the isotropy property as well
inherits the isotropy property as well ![\expect[xx^\top] = I](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-693de07bc613a6824b8000f4615ccf85_l3.png "Rendered by QuickLaTeX.com") . As a consequence, we can use the vector
. As a consequence, we can use the vector ![\expect[x^\top Ax] = \tr(A)](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-99c590c818d205b76d1b6ab2b4391edb_l3.png "Rendered by QuickLaTeX.com") . Trace estimation has been a frequent topic
. Trace estimation has been a frequent topic  ? This question
? This question  is
is ![\[\Var(x^\top A x) = \expect[(x^\top A x)^2] - \expect[x^\top A x]^2 = \expect[(x^\top A x)^2] - \tr(A)^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5647f8054d7455519ae8e1bef31af139_l3.png "Rendered by QuickLaTeX.com")
![\expect[(x^\top A x)^2]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b5fbf8e67e4ff29170a0eebe078ce961_l3.png "Rendered by QuickLaTeX.com") . Suppose that the individual base vectors
. Suppose that the individual base vectors ![\[\expect[(x_i^\top A x_i)^2] \le \alpha \tr(A)^2 \quad \text{for every psd matrix } A.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-277e90ec9bcf7846732486a3c5953375_l3.png "Rendered by QuickLaTeX.com")
 . Under assumption (1), Meyer and Avron show that the tensor-product trace estimator
. Under assumption (1), Meyer and Avron show that the tensor-product trace estimator ![\[\expect[(x^\top A x)^2] \le \alpha^\ell \tr(A)^2 \quad \text{for any psd matrix } A.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ee2f7f4328d1d2f7117f850c28a62528_l3.png "Rendered by QuickLaTeX.com")
 for any vector
for any vector  . Observe, the constant in the bound (2) is exponentially larger than the constant in bound (1). Unfortunately,
. Observe, the constant in the bound (2) is exponentially larger than the constant in bound (1). Unfortunately,  is a tensor product of isotropic vectors
is a tensor product of isotropic vectors  and
and  , and suppose that
, and suppose that ![\[\expect[(y^\top A_1 y)^2] \le \alpha_1 \tr(A_1)^2 \quad \text{and} \quad \expect[(z^\top A_2 z)^2] \le \alpha_2 \tr(A_2)^2 \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9b8a7bf17b8f16b0ec73e0027423055e_l3.png "Rendered by QuickLaTeX.com")
 and
and  . Now, let
. Now, let  psd matrix, and
psd matrix, and ![\[A = \begin{bmatrix} A_{11} & \cdots & A_{1d_1} \\ \vdots & \ddots & \vdots \\ A_{d_1 1} & \cdots & A_{d_1 d_1} \end{bmatrix}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-43bb38960318d2dc04d04e021243f074_l3.png "Rendered by QuickLaTeX.com")

 alone
alone![\begin{align*}\expect_y [(x^\top A x)^2] &\le \alpha_1 \left( \tr \begin{bmatrix} z^\top A_{11}z & \cdots & z^\top A_{1d_1}z \\ \vdots & \ddots & \vdots \\ z^\top A_{d_1 1}z & \cdots & z^\top A_{d_1 d_1}z \end{bmatrix} \right)^2 \\ &= \alpha_1 [z^\top(A_{11}+\cdots+ A_{d_1d_1})z]^2.\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4536f123e88a9b329362cc19825464cc_l3.png "Rendered by QuickLaTeX.com")
 and invoking
and invoking ![\begin{align*}\expect [(x^\top A x)^2] &\le \alpha_1 \alpha_2 \tr(A_{11}+\cdots+ A_{d_1d_1})^2=\alpha_1 \alpha_2\tr(A)^2.\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5f49a9eb10fd37e189a8f518add54828_l3.png "Rendered by QuickLaTeX.com")
 . Voilà! We have deduced the bound
. Voilà! We have deduced the bound ![\[\expect [(x^\top A x)^2] \le \alpha_1\alpha_2 \tr(A)^2,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-434b9be6486b07cf1509bbe664e7f3d4_l3.png "Rendered by QuickLaTeX.com")

![[-1,1]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-634fe4705a526f221e312cffd3ceda93_l3.png "Rendered by QuickLaTeX.com") into
into ![\[\max_{x \in [-1,1]} |p'(x)| \le n^2. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-63dfe56d2bb34f3fc695f743fdf67b40_l3.png "Rendered by QuickLaTeX.com")
![[-1,1] \times [-1,1]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1c563b2ce110b55706b82573366be7c8_l3.png "Rendered by QuickLaTeX.com") , the fastest it can wiggle—as measured by its first
, the fastest it can wiggle—as measured by its first  . This function maps
. This function maps ![\[\max_{x \in [-1,1]} |p'(x)| = \max_{x \in [-1,1]} |nx^{n-1}| = n\ll n^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-fac35ca5506dda4220ce3027702d9f37_l3.png "Rendered by QuickLaTeX.com")
 , which are motivated and described at length in
, which are motivated and described at length in  (orange dashed line).
(orange dashed line).![\[\max_{x \in [-1,1]} |T_n'(x)| = n^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a5f7b0314564d6e87a09ef03981df060_l3.png "Rendered by QuickLaTeX.com")
 .
.![\[\max_{x \in [-1,1]} |p^{(k)}(x)| \le \max_{x \in [-1,1]} |T_d^{(k)}| = \frac{n^2(n^2-1^2)\cdots(n^2-(k-1)^2)}{1\times 3 \times \cdots \times (2k-1)}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-625bc4245d3cc1d47a534e8d824d9a9a_l3.png "Rendered by QuickLaTeX.com")
 . The inequality (2) is often called the
. The inequality (2) is often called the ![[a,b] \times [c,d]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4c5eee6ce9a02aaea141b2b44fa5d05e_l3.png "Rendered by QuickLaTeX.com") of general sidelengths. Indeed, any polynomial
of general sidelengths. Indeed, any polynomial ![[a,b]\to[c,d]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2a7815349e537249459ea754a1bc3744_l3.png "Rendered by QuickLaTeX.com") can be transmuted to a polynomial
can be transmuted to a polynomial  mapping
mapping ![\[\tilde{p}(x) = -1 + 2\cdot \frac{p(\ell(x)) - c}{d-c} \quad \text{for } \ell(x) = a + (b-a) \cdot \frac{x+1}{2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3f013f343441b1f0556a72513ea33d9e_l3.png "Rendered by QuickLaTeX.com")
 maps
maps ![\[\max_{t \in [a,b]} |p'(t)| = \frac{d-c}{b-a} \cdot \max_{x \in [-1,1]} |\tilde p'(x)|.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-20e2a9e85c0f8c7703ec8843c61ddfa8_l3.png "Rendered by QuickLaTeX.com")
![[a,b]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-336bb22d4b2092329486008f6665b888_l3.png "Rendered by QuickLaTeX.com") to
to ![[c,d]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9a98449615ceb53e7851488b33334426_l3.png "Rendered by QuickLaTeX.com") . Then
. Then ![\[\max_{t \in [a,b]} |p'(t)| \le \frac{d-c}{b-a} \cdot n^2. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-752b97244cedaa80b3c201ab0d27ac8c_l3.png "Rendered by QuickLaTeX.com")
 ,
,  , or
, or  by a polynomial. There are
by a polynomial. There are  “. That is, we seek lower bounds on the polynomial needed to approximate a given function to a specified level of accuracy.
“. That is, we seek lower bounds on the polynomial needed to approximate a given function to a specified level of accuracy.![[0,b]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-fed604a8d666b6b271118b6980e2dd5e_l3.png "Rendered by QuickLaTeX.com") ?
? . To address this question, suppose that
. To address this question, suppose that ![\[|p(t) - \e^{-t}| \le 0.1 \quad \text{for all }t \in [0,b].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e47cf528e4b9076d406c0fdbb28dd879_l3.png "Rendered by QuickLaTeX.com")
 for positive
for positive  , it therefore must hold that
, it therefore must hold that  . Therefore, the polynomial
. Therefore, the polynomial ![[0,b] \times [-0.1,1.1]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1fc744b538cdc16ff0989123627d98fd_l3.png "Rendered by QuickLaTeX.com") .
. decreases quite rapidly. At zero, it is
decreases quite rapidly. At zero, it is  and at two, it is
and at two, it is  . Therefore, since
. Therefore, since  is within
is within  is at least
is at least  and
and  is at most
is at most  . Therefore, by the intermediate value theorem, there is some
. Therefore, by the intermediate value theorem, there is some  between
between  and
and  for which
for which ![\[p'(t^*) = \frac{p(2) - p(0)}{2-0} \ge \frac{0.9 - 0.24}{2}=0.33.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-128d3240b1f7024150800ae2aea13002_l3.png "Rendered by QuickLaTeX.com")
![\[0.33 \le p'(t^*) \le \max_{t \in [0,b]} |p'(t)| \le \frac{1.1-(-0.1)}{b-0} \cdot n^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5abd7df66a224f73f72aeca9680f0d92_l3.png "Rendered by QuickLaTeX.com")
![\[n \ge \sqrt{\frac{0.33}{1.2}} \cdot \sqrt{b} > 0.5 \sqrt{b}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0aa42a6b5c1734de97fe13b82c211538_l3.png "Rendered by QuickLaTeX.com")
 to approximate
to approximate  also suffices to approximate
also suffices to approximate  and
and  . For a bit of fun, see if you can get results for approximating a power
. For a bit of fun, see if you can get results for approximating a power  on
on  . You may be surprised by what you find!
. You may be surprised by what you find! is necessary for approximating the ramp function
is necessary for approximating the ramp function ![\[f_{\rm ramp}(t) = \begin{cases} 1-t/2, & 0 \le t \le 2, \\ 0, & 2 < t \le b.\end{cases}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-26e453856802dfaca0eef8e4b383a649_l3.png "Rendered by QuickLaTeX.com")
 converge at the rate
converge at the rate  . But in terms of the polynomial degree to start approximating these functions, both functions require the same degree
. But in terms of the polynomial degree to start approximating these functions, both functions require the same degree  . Pretty neat, I think.
. Pretty neat, I think.
 , but these results give poor results for intermediate block sizes
, but these results give poor results for intermediate block sizes  . But
. But  . Check out
. Check out ![\[K = \begin{bmatrix} G & AG & \cdots & A^{t-1}G \end{bmatrix},\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3a6ec37fe54d09f2ee9f6f2c648dafd1_l3.png "Rendered by QuickLaTeX.com")
 real
real  is an
is an  random
random ![\[p_a(u) = a_1 + a_2 u+ \cdots + a_t u^{t-1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ba898ff67858e9c5a76463593337b542_l3.png "Rendered by QuickLaTeX.com")
 and have written the polynomial
and have written the polynomial  parametrically in terms of its vector of coefficients
parametrically in terms of its vector of coefficients  . The polynomials of degree at most
. The polynomials of degree at most  provide a
provide a  to be complex numbers.
to be complex numbers.
 be
be  (distinct) input locations. If we evaluate
(distinct) input locations. If we evaluate  at each number, we obtain a list of (output) values, which we denote by
at each number, we obtain a list of (output) values, which we denote by  of
of ![\[p_a(\lambda_i) = \sum_{j=1}^t \lambda_i^{j-1} a_j.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-eaaf53acb1f30e1ced80af4704800e02_l3.png "Rendered by QuickLaTeX.com")
 are a nonlinear function of the input values
are a nonlinear function of the input values  but a linear function of the coefficients
but a linear function of the coefficients  . We may call the mapping
. We may call the mapping  the coefficients-to-values map.
the coefficients-to-values map.
 . It is given by the formula
. It is given by the formula ![\[V= \begin{bmatrix} 1 & \lambda_1 & \lambda_1^2 & \cdots & \lambda_1^{t-1} \\ 1 & \lambda_2 & \lambda_2^2 & \cdots & \lambda_2^{t-1} \\ 1 & \lambda_3 & \lambda_3^2 & \cdots & \lambda_3^{t-1} \\ \vdots & \vdots & \vdots & \ddots & \vdots \\ 1 & \lambda_s & \lambda_s^2 & \cdots & \lambda_s^{t-1} \end{bmatrix} \in \complex^{s\times t}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2d100f803f43a4910ba6c03d2edde384_l3.png "Rendered by QuickLaTeX.com")
 .
.
 so the number of locations
so the number of locations  equals the number of coefficients
equals the number of coefficients  . Its inverse
. Its inverse  maps a set of values
maps a set of values ![\[p_a(\lambda_i) = f_i \quad \text{for } i =1,\ldots,t .\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0cab3d96a2006c60b95e665b3c9afc97_l3.png "Rendered by QuickLaTeX.com")
 , obtaining a vector of coefficients
, obtaining a vector of coefficients  . Then, define the interpolating polynomial
. Then, define the interpolating polynomial ![\[q(u) = a_1 + a_2 u + \cdots + a_t u^{t-1} \quad \text{with } a = V^{-1} f. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b44af0dbb2f4e6b3c74a40b23d074f68_l3.png "Rendered by QuickLaTeX.com")
 interpolates the values
interpolates the values  ,
,  .
.
 that is zero at the locations
that is zero at the locations  but nonzero at the first location
but nonzero at the first location  . (Remember that we have assumed that
. (Remember that we have assumed that  are distinct.) Further, by rescaling this polynomial to
are distinct.) Further, by rescaling this polynomial to![\[\ell_1(u) = \frac{(u - \lambda_2)(u - \lambda_3)\cdots(u-\lambda_t)}{(\lambda_1 - \lambda_2)(\lambda_1 - \lambda_3)\cdots(\lambda_1-\lambda_t)},\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-174996c439ec13555388925748a3269e_l3.png "Rendered by QuickLaTeX.com")
 , we can define a similar polynomial
, we can define a similar polynomial ![\[\ell_i(u) = \frac{(u - \lambda_1)\cdots(u-\lambda_{i-1}) (u-\lambda_{i+1})\cdots(u-\lambda_t)}{(\lambda_i - \lambda_1)\cdots(\lambda_i-\lambda_{i-1}) (\lambda_i-\lambda_{i+1})\cdots(\lambda_i-\lambda_t)}, \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5453dea91bda2e744ca98b5dc0ae9790_l3.png "Rendered by QuickLaTeX.com")
 for
for  . Using the Dirac delta symbol, we may write
. Using the Dirac delta symbol, we may write ![\[\ell_i(\lambda_j) = \delta_{ij} = \begin{cases} 1, & i = j, \\ 0, & i\ne j. \end{cases}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-64c4effeb0f12225a76059d89ccce692_l3.png "Rendered by QuickLaTeX.com")
 are called the
are called the  . Below is an interactive illustration of the second Lagrange polynomial
. Below is an interactive illustration of the second Lagrange polynomial  associated with the points
associated with the points  (with
(with  ).
).
 and sum up, obtaining
and sum up, obtaining ![\[q(u) = \sum_{i=1}^t f_i \ell_i(u).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c1c5043718b5df795a883a6a60d26aaf_l3.png "Rendered by QuickLaTeX.com")
![\[q(\lambda_j) = \sum_{i=1}^t f_i \ell_i(\lambda_j) = \sum_{i=1}^t f_i \delta_{ij} = f_j. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-90032b377ebe250ef36dccba2186b6ce_l3.png "Rendered by QuickLaTeX.com")
 . To convert between these formulas, we just need to express the Lagrange polynomial basis in the monomial basis.
. To convert between these formulas, we just need to express the Lagrange polynomial basis in the monomial basis. , and consider the fourth unnormalized Lagrange polynomial
, and consider the fourth unnormalized Lagrange polynomial ![\[(u - \lambda_1) (u - \lambda_2) (u - \lambda_3).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d3fe2471c7c0557476992f6f7a1361cf_l3.png "Rendered by QuickLaTeX.com")
 , we obtain the expression
, we obtain the expression ![\[(u - \lambda_1) (u - \lambda_2) (u - \lambda_3) = u^3 - (\lambda_1 + \lambda_2 + \lambda_3) u^2 + (\lambda_1\lambda_2 + \lambda_1\lambda_3 + \lambda_2\lambda_3)u - \lambda_1\lambda_2\lambda_3.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ad4d01202c2b9a69e395412829cc7fed_l3.png "Rendered by QuickLaTeX.com")
 , we recognize some pretty distinctive expressions involving the
, we recognize some pretty distinctive expressions involving the 
 , the
, the  is defined as the sum of all products
is defined as the sum of all products  of
of ![\[e_k(\mu_1,\ldots,\mu_{t-1}) = \sum_{i_1 < i_2 < \cdots < i_k} \mu_{i_1}\mu_{i_2}\cdots \mu_{i_k}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-49b00bf8f1a0f6fc399e029516a4682e_l3.png "Rendered by QuickLaTeX.com")
 :
:![\[(u + \mu_1) (u + \mu_2) \cdots (u+\mu_k) = \sum_{j=0}^k e_{k-j}(\mu_1,\ldots,\mu_k) u^j.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-fe26dd7ba2e215fba4fea5cd28147cb2_l3.png "Rendered by QuickLaTeX.com")
 denote the list of locations without
denote the list of locations without ![\[\ell_i(u) = \frac{\sum_{j=1}^t e_{t-j}(-\lambda_{-i})u^{j-1}}{\prod_{k\ne i} (\lambda_i - \lambda_k)}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-15d6cb602dcf916e14363b25a013870d_l3.png "Rendered by QuickLaTeX.com")
![\[q(u) = \sum_{i=1}^t f_i\ell_i(u) = \sum_{i=1}^t \sum_{j=1}^t f_i \frac{e_{t-j}(-\lambda_{-i})}{\prod_{k\ne i} (\lambda_i - \lambda_k)} u^{j-1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-17012971c8949a309b08ae230bf7fa94_l3.png "Rendered by QuickLaTeX.com")
![\[q(u) = \sum_{j=1}^t \sum_{i=1}^t f_i \frac{e_{t-j}(-\lambda_{-i})}{\prod_{k\ne i} (\lambda_i - \lambda_k)} u^{j-1} = \sum_{j=1}^t \left(\sum_{i=1}^t \frac{e_{t-j}(-\lambda_{-i})}{\prod_{k\ne i} (\lambda_i - \lambda_k)}\cdot f_i \right) u^{j-1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-10960bce48c6a124c253db4c1272f2e2_l3.png "Rendered by QuickLaTeX.com")
![\[a_j = \sum_{i=1}^t \frac{e_{t-j}(-\lambda_{-i})}{\prod_{k\ne i} (\lambda_i - \lambda_k)}\cdot f_i.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2bb3d1280c99ca124084f031ed343679_l3.png "Rendered by QuickLaTeX.com")
![\[(V^{-1})_{ji} = \frac{e_{t-j}(-\lambda_{-i})}{\prod_{k\ne i} (\lambda_i - \lambda_k)}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c3d618b9ee7195d040d1875f76fb1d67_l3.png "Rendered by QuickLaTeX.com")
 , the condition number of
, the condition number of ![\[\kappa_{\uinorm{\cdot}}(V) = \uinorm{V} \uinorm{\smash{V^{-1}}}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-cf4da36f5f2ce121d2d2512423310221_l3.png "Rendered by QuickLaTeX.com")
 is chosen to be the (
is chosen to be the (![\[\norm{A}_1= \max_{x \ne 0} \frac{\norm{Ax}_1}{\norm{x}_1} \quad \text{where } \norm{x}_1 = \sum_i |x_i|.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-beb68f2e11bc5c6e30b06fec3b886ff3_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{A}_\infty = \max_j \sum_i |A_{ij}|. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-936a7309e99d6c03a128b3a7771c88df_l3.png "Rendered by QuickLaTeX.com")
 .
.
 is straightforward. Indeed, setting
is straightforward. Indeed, setting  and using (5), we compute
and using (5), we compute ![\[\norm{V}_1= \max_{1\le j \le t} \sum_{i=1}^t |\lambda_i|^{j-1} \le \max_{1\le j \le t} tM^{j-1} = t\max\{1,M^{t-1}\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ec5f488cb8233df69dd22a00e9bae784_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{V}_1\le t(1+M)^{t-1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8cecac332b3fcbe899304e963ec1fe47_l3.png "Rendered by QuickLaTeX.com")
 . Fortunately, we have already done most of the hard work needed to bound this quantity. Using our expression (4) for the entries of
. Fortunately, we have already done most of the hard work needed to bound this quantity. Using our expression (4) for the entries of ![\[\norm{\smash{V^{-1}}}_1= \max_{1\le j \le t} \sum_{i=1}^t |(V^{-1})_{ij}| = \max_{1\le j \le t}\frac{ \sum_{i=1}^t |e_{t-i}(-\lambda_{-j})|}{\prod_{k\ne j} |\lambda_j- \lambda_k|}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b431915bf4dd9e0a3087cd91f5e3533a_l3.png "Rendered by QuickLaTeX.com")
![\[|e_k(\mu_1,\ldots,\mu_s)| \le e_k(|\mu_1|,\ldots,|\mu_s|).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-73a7d10b6aadf0271cb538d648049f13_l3.png "Rendered by QuickLaTeX.com")
 , we obtain
, we obtain![\[\norm{\smash{V^{-1}}}_1\le \max_{1\le j \le t}\frac{ \sum_{i=1}^t e_{t-i}(|\lambda_{-j}|)}{\prod_{k\ne j} |\lambda_j- \lambda_k|}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-34c4b502a43c91513781e51c25190ad1_l3.png "Rendered by QuickLaTeX.com")
 to obtain the expression
to obtain the expression ![\[\norm{\smash{V^{-1}}}_1\le \max_{1\le j \le t}\frac{ \prod_{k\ne j} (1 + |\lambda_k|)}{\prod_{k\ne j} |\lambda_j- \lambda_k|}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9fee19675094e57026eaf8571e060317_l3.png "Rendered by QuickLaTeX.com")
 . To bound the denominator, let
. To bound the denominator, let  be the smallest distance between two locations. Using
be the smallest distance between two locations. Using  and
and  , we can weaken the bound (6) to obtain
, we can weaken the bound (6) to obtain ![\[\norm{\smash{V^{-1}}}_1\le \left(\frac{1+M}{\mathrm{gap}}\right)^{t-1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4df225e8dea0e69df68907fc81ab32e6_l3.png "Rendered by QuickLaTeX.com")
 from above, we obtain a bound on the condition number
from above, we obtain a bound on the condition number ![\[\kappa_1(V) \le t \left( \frac{(1+M)^2}{\mathrm{gap}} \right)^{t-1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a88bac288484194059179a7e854bc368_l3.png "Rendered by QuickLaTeX.com")
 . Then
. Then ![\[\norm{\smash{V^{-1}}}_1\le \left(\frac{1+M}{\mathrm{gap}}\right)^{t-1}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-933010efafd6e0336cdf5b4071c2c90b_l3.png "Rendered by QuickLaTeX.com")
 polynomial has precisely
polynomial has precisely  of the values
of the values  ? Well, if we set all the coefficients
? Well, if we set all the coefficients  to zero, then
to zero, then  as well. So to avoid this trivial case, we should enforce a normalization condition on the coefficient vector
as well. So to avoid this trivial case, we should enforce a normalization condition on the coefficient vector  . With this setting, we are ready to compute. Begin by observing that
. With this setting, we are ready to compute. Begin by observing that ![\[\min_{\norm{a}_1 = 1} \norm{Va}_1 = \min_{a\ne 0} \frac{\norm{Va}_1}{\norm{a}_1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3a21773d318eeb447cc3deec7d2ec9e1_l3.png "Rendered by QuickLaTeX.com")
 , obtaining
, obtaining ![\[\min_{\norm{a}_1 = 1} \norm{Va}_1 = \min_{a\ne 0} \frac{\norm{Va}_1}{\norm{a}_1} = \min_{f\ne 0} \frac{\norm{f}_1}{\norm{\smash{V^{-1}f}}_1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e223271ff5f99accd84933f4936864bd_l3.png "Rendered by QuickLaTeX.com")
![\[\left(\min_{\norm{a}_1 = 1} \norm{Va}_1\right)^{-1} = \max_{f\ne 0} \frac{\norm{\smash{V^{-1}f}}_1}{\norm{f}_1} = \norm{\smash{V^{-1}}}_1.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-745fdb308eb43cd71eb1fda9a7b16762_l3.png "Rendered by QuickLaTeX.com")
![\[\min_{\norm{a}_1 = 1} \norm{Va}_1 = \norm{\smash{V^{-1}}}_1^{-1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-145e10d104c261b8eba6886d9e5a88d0_l3.png "Rendered by QuickLaTeX.com")
![\[\min_{\uinorm{v} = 1} \uinorm{Av} = \min_{v\ne 0} \frac{\uinorm{Av}}{\uinorm{v}} = \uinorm{\smash{A^{-1}}}^{-1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c07d1dd4195814db2ab892e8587fb9b4_l3.png "Rendered by QuickLaTeX.com")
 on the values
on the values  and locations
and locations ![\[|p(\lambda_1)| + \cdots + |p(\lambda_t)| \ge \left(\frac{\mathrm{gap}}{1+M}\right)^{t-1} (|a_1| + \cdots + |a_t|).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f011798a0af29d677a21bf1d0fb00152_l3.png "Rendered by QuickLaTeX.com")
![\expect[zf(z)] = \expect[f'(z)]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7dba10c0a29b0f68eefa135a0e67d0cb_l3.png "Rendered by QuickLaTeX.com") .
.![\expect[z^2]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b789f7d051bbf792fea37f44f88b1f83_l3.png "Rendered by QuickLaTeX.com") of a standard Gaussian random variable
of a standard Gaussian random variable  to obtain
to obtain ![\[\expect[z^2] = \expect[z f(z)] = \expect[f'(z)] = \expect[1] = 1.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-72017e2492468cdba39e4878b12f76aa_l3.png "Rendered by QuickLaTeX.com")
 , we compute
, we compute ![\[\expect[z^4] = \expect[zf(z)] = \expect[f'(z)] = 3\expect[z^2] = 3.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d7582a91dd80de04d4bb3bd17c2969cd_l3.png "Rendered by QuickLaTeX.com")
![\[\expect[z^{2p}] = \expect[z\cdot z^{2p-1}] = (2p-1) \expect[z^{2p-2}] = (2p-1)(2p-3) \expect[z^{2p-4}] = \cdots = (2p-1)(2p-3)\cdots 1.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-463cff05d7ce30d54b9325a0b4d32da8_l3.png "Rendered by QuickLaTeX.com")
 th moment of a standard Gaussian random variable is
th moment of a standard Gaussian random variable is  , where
, where  indicates the (in)famous
indicates the (in)famous  .
.
![\expect[|z|]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8dcb87553abe8c547e97479e4ef0ad22_l3.png "Rendered by QuickLaTeX.com") . To do so, we choose
. To do so, we choose  to be the
to be the ![\[\operatorname{sign}(x) = \begin{cases} 1, & x > 0, \\ 0 & x = 0, \\ -1 & x < 0.\end{cases}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-28ef3c2b8f17f4fc65f112fc1ea06c86_l3.png "Rendered by QuickLaTeX.com")
 , where
, where  denotes the famous
denotes the famous ![\[\expect[|z|] = \expect[zf(z)] = \expect[f'(z)] = 2\expect[\delta(z)].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ff50b28c78f8b8d69843835ac3bd7fab_l3.png "Rendered by QuickLaTeX.com")
![\expect[\delta(z)]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-078369af4f64b86ff6fe4d075710549d_l3.png "Rendered by QuickLaTeX.com") , write the integral out using the
, write the integral out using the  of the standard Gaussian distribution:
of the standard Gaussian distribution: ![\[\expect[|z|] = 2\expect[\delta(z)] = 2\int_{-\infty}^\infty \phi(x) \delta(x) \, \mathrm{d} x = 2\phi(0).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-96eedb8599b918aed9f53f71548b47d8_l3.png "Rendered by QuickLaTeX.com")
![\[\phi(x) = \frac{1}{\sqrt{2\pi}} \exp \left( - \frac{x^2}{2} \right),\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-92edc4a207255caae1eedb17381a807f_l3.png "Rendered by QuickLaTeX.com")
![\[\expect[|z|] = 2 \cdot \frac{1}{\sqrt{2 \pi}} = \sqrt{\frac{2}{\pi}}}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d1d32c2b5642230b0f179641bf6b5a65_l3.png "Rendered by QuickLaTeX.com")
 denote the eigenvalues of
denote the eigenvalues of  .
.  . After many iterations,
. After many iterations,  denote the initial vector, the
denote the initial vector, the  and the
and the ![\[\mu^{(t)} = \left(x^{(t)}\right)^\top Ax^{(t)}= \frac{\left(x^{(0)}\right)^\top A^{2t+1} x^{(0)}}{\left(x^{(0)}\right)^\top A^{2t} x^{(0)}}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8744da7cad08a86191f52369bd86e7b3_l3.png "Rendered by QuickLaTeX.com")
 of
of ![\[\mu^{(t)} = \frac{\sum_{i=1}^n \lambda_i^{2t+1} z_i^2}{\sum_{i=1}^n \lambda_i^{2t} z_i^2},\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-91c240175a5862804e47c506606be709_l3.png "Rendered by QuickLaTeX.com")
 as an approximation to the dominant eigenvalue
as an approximation to the dominant eigenvalue ![\[\frac{\lambda_1 - \mu^{(t)}}{\lambda_1} = \frac{\sum_{i=1}^n (\lambda_1 - \lambda_i)/\lambda_1 \cdot \lambda_i^{2t} z_i^2}{\sum_{i=1}^n \lambda_i^{2t} z_i^2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-667bb71dd8089d5b26d87d625b82e884_l3.png "Rendered by QuickLaTeX.com")
![\[\frac{\lambda_1 - \lambda_i}{\lambda_1}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0a300e8efbe358c2e744445cb4d0c552_l3.png "Rendered by QuickLaTeX.com")
 and is at most one for
and is at most one for  . Therefore, the error is bounded as
. Therefore, the error is bounded as ![\[\frac{\lambda_1 - \mu^{(t)}}{\lambda_1} \le \frac{\sum_{i=2}^n \lambda_i^{2t} z_i^2}{\lambda_1^{2t} z_1^2 + \sum_{i=2}^n \lambda_i^{2t} z_i^2} = \frac{c^2}{\lambda_1^{2t} z_1^2 + c^2} \quad \text{for } c^2 = \sum_{i=2}^n \lambda_i^{2t} z_i^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-752c7ced1eb1bd8cc0ddf1eacdbc5b1c_l3.png "Rendered by QuickLaTeX.com")
 to consolidate the terms in this expression not depending on
to consolidate the terms in this expression not depending on  . Since
. Since  with respect to only the randomness in the first Gaussian variable
with respect to only the randomness in the first Gaussian variable ![\[f'(x) = \frac{c^2}{\lambda_1^{2t}x^2 + c^2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5a451acfc5ab3bdec6db9fab5c32cf9b_l3.png "Rendered by QuickLaTeX.com")
![\[f(x) = \frac{c}{\lambda_1^t} \arctan \left( \frac{\lambda_1^t}{c} \cdot x \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6e54f3861046933630d2185bac2fb159_l3.png "Rendered by QuickLaTeX.com")
![\[\expect_{z_1}\left[\frac{\lambda_1 - \mu^{(t)}}{\lambda_1}\right] \le \expect_{z_1} \left[z_1 \cdot \frac{c}{\lambda_1} \arctan \left( \frac{\lambda_1^t}{c} \cdot z_1 \right) \right] = \expect_{z_1} \left[|z_1| \cdot \frac{c}{\lambda_1^t} \left|\arctan \left( \frac{\lambda_1^t}{c} \cdot z_1 \right)\right| \right].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b8e649fd7a63c495f6f2fa09d9534c3b_l3.png "Rendered by QuickLaTeX.com")
 , so we can bound
, so we can bound![\[\expect_{z_1}\left[\frac{\lambda_1 - \mu^{(t)}}{\lambda_1}\right] \le = \frac{\pi}{2} \cdot \frac{c}{\lambda_1^t} \cdot \expect_{z_1} \left[|z_1|\right] \le \sqrt{\frac{\pi}{2}} \cdot \frac{c}{\lambda_1^t}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e9d84cb3710fdc80b0166f5491e5b3da_l3.png "Rendered by QuickLaTeX.com")
![\expect[|z|] = \sqrt{2/\pi}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-72fae41f864e69e247552190c6b1ef2e_l3.png "Rendered by QuickLaTeX.com") for a standard Gaussian variable
for a standard Gaussian variable ![\[c = \sqrt{\sum_{i=2} \lambda_i^{2t} z_i^2} \le \lambda_2^{t} \sqrt{\sum_{i=2}^n z_i^2} = \lambda_2^t \norm{(z_2,\ldots,z_n)}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7facff60e6dd9b6579d16ed28a6b0f47_l3.png "Rendered by QuickLaTeX.com")
 times the length of a vector of
times the length of a vector of  standard Gaussian entries. As we’ve
standard Gaussian entries. As we’ve  . Thus,
. Thus, ![\expect[c] \le \lambda_2^t \sqrt{n-1}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-334812a1686dc037c03bd91572144ee7_l3.png "Rendered by QuickLaTeX.com") . We conclude that the expected error for power iteration is
. We conclude that the expected error for power iteration is ![\[\expect\left[\frac{\lambda_1 - \mu^{(t)}}{\lambda_1}\right] = \expect_c\left[\expect_{z_1} \left[ \frac{\lambda_1 - \mu^{(t)}}{\lambda_1} \right]\right] \le \sqrt{\frac{\pi}{2}} \cdot \expect\left[ \frac{c}{\lambda_1^t} \right] =\sqrt{\frac{\pi}{2}} \cdot \left(\frac{\lambda_2}{\lambda_1}\right)^t \cdot \sqrt{n-1} .\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-71e78c2a3b2280f94d13d08cfeed954c_l3.png "Rendered by QuickLaTeX.com")
 .
.
![\[\text{Find $x$ such that } Ax = b. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-29d9a4727a905120c7e471f5f6d88048_l3.png "Rendered by QuickLaTeX.com")
 . Beginning from an initial iterate
. Beginning from an initial iterate  , randomized Kaczmarz works as follows. For
, randomized Kaczmarz works as follows. For  :
:
 with probability
with probability  .
. holds exactly:
holds exactly: ![\[x_{t+1} \coloneqq x_t + \frac{b_{i_t} - a_{i_t}^\top x_t}{\norm{a_{i_t}}^2} a_{i_t}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-99cc99e6de2ca367d24ee6f20e610735_l3.png "Rendered by QuickLaTeX.com")
 denotes the
denotes the  th row of
th row of  should we use? The answer to this question may depend on whether the system (1) is
should we use? The answer to this question may depend on whether the system (1) is ![\[p_j = \frac{\norm{a_j}^2}{\norm{A}_{\rm F}^2} \quad \text{for } j = 1,2,\ldots,n. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c0282178b77487afd710ad66bb50707b_l3.png "Rendered by QuickLaTeX.com")
![\[p_j = \frac{1}{n} \quad \text{for } j = 1,2,\ldots,n. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b0a9325d8b121aaab8d4547fa378ffed_l3.png "Rendered by QuickLaTeX.com")
 with entries
with entries  , and choose the right-hand side
, and choose the right-hand side  with standard Gaussian random entries. The convergence of standard RK with sampling rule (1) and uniform RK with sampling rule (2) is shown in the plot below. After a million iterations, the difference in final accuracy is dramatic: the final relative error 0.00012 was uniform RK and 0.67 for standard RK!
with standard Gaussian random entries. The convergence of standard RK with sampling rule (1) and uniform RK with sampling rule (2) is shown in the plot below. After a million iterations, the difference in final accuracy is dramatic: the final relative error 0.00012 was uniform RK and 0.67 for standard RK!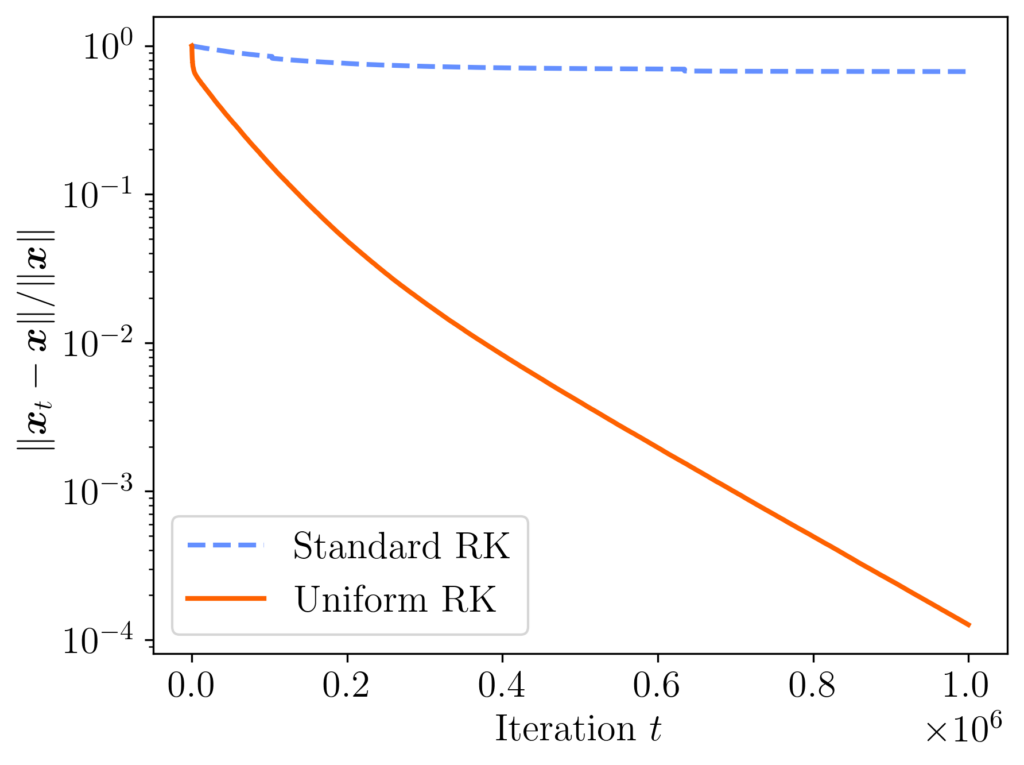
 , then the performance of both methods is similar, with both methods ending with a relative error of about 0.07.
, then the performance of both methods is similar, with both methods ending with a relative error of about 0.07.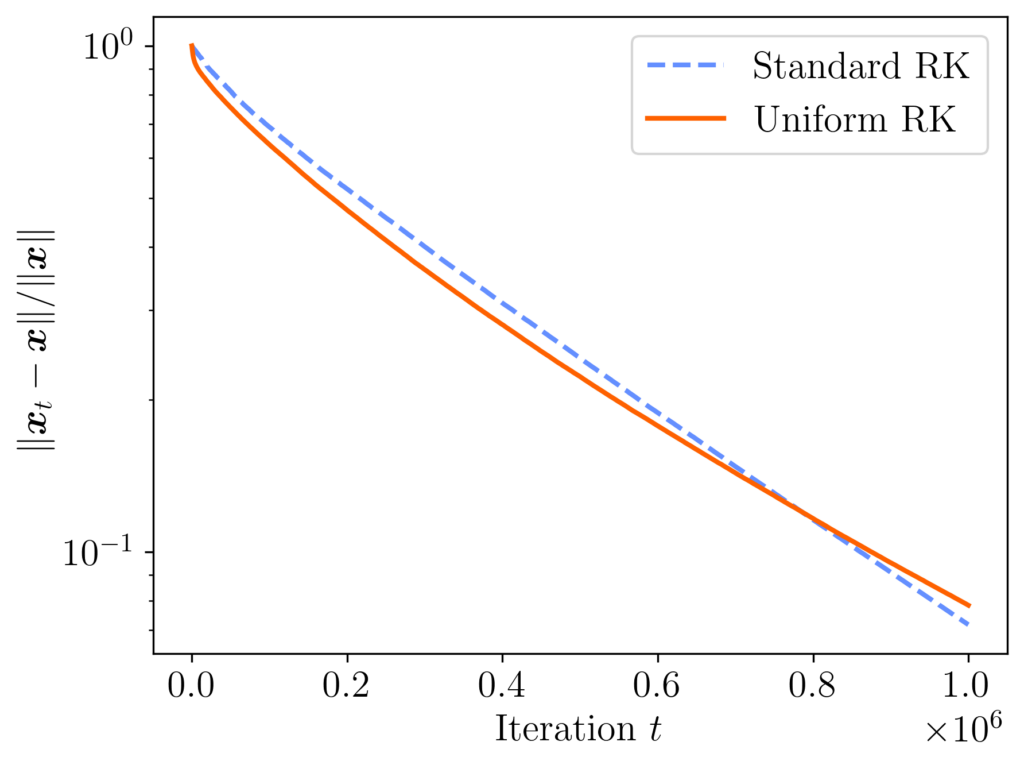
![\[\expect\left[ \norm{x_t - x_\star}^2 \right] \le (1 - \kappa_{\rm dem}(A)^{-2})^t \norm{x_\star}^2. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1f788737bb459ed2beaab020bca517c2_l3.png "Rendered by QuickLaTeX.com")
![\[\kappa_{\rm dem}(A) = \frac{\norm{A}_{\rm F}}{\sigma_{\rm min}(A)} = \sqrt{\sum_i \left(\frac{\sigma_i(A)}{\sigma_{\rm min}(A)}\right)^2} \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-824557fbf90de5be0f9756f9089728aa_l3.png "Rendered by QuickLaTeX.com")
 are the
are the  is consistent, possessing a solution
is consistent, possessing a solution  . If there are multiple solutions, we let
. If there are multiple solutions, we let  denote a diagonal matrix containing the inverse row norms, and introduce the row-equilibrated matrix
denote a diagonal matrix containing the inverse row norms, and introduce the row-equilibrated matrix  . The row-equilibrated matrix
. The row-equilibrated matrix  as standard RK applied to the row-equilibrated system
as standard RK applied to the row-equilibrated system  .
.![\[\expect\left[ \norm{\hat{x}_t - x_\star}^2 \right] \le (1 - \kappa_{\rm dem}(D_A A)^{-2})^t \norm{x_\star}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3fc6402842af70a33954d0133066a5e3_l3.png "Rendered by QuickLaTeX.com")
![\[\kappa(A) \coloneqq \frac{\sigma_{\rm max}(A)}{\sigma_{\rm min}(A)}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3fb2bba4d99c34bc055f7043fccfe19c_l3.png "Rendered by QuickLaTeX.com")
 . Also, let
. Also, let  denote the set of all (nonsingular) diagonal matrices.
denote the set of all (nonsingular) diagonal matrices.
 be wide
be wide  and full-rank, and let
and full-rank, and let  denote the row-scaling of
denote the row-scaling of ![\[\kappa_{\rm dem}(D_AA) \le \sqrt{n}\cdot \min_{D \in \mathrm{Diag}} \kappa (DA) \le \sqrt{n}\cdot \min_{D \in \mathrm{Diag}} \kappa_{\rm dem} (DA).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ec117b2c8c81cbcfc88eeae87eac0aaa_l3.png "Rendered by QuickLaTeX.com")
 , this result shows that implementing randomized Kaczmarz with uniform sampling yields to a convergence rate that is within a factor of
, this result shows that implementing randomized Kaczmarz with uniform sampling yields to a convergence rate that is within a factor of  denote the maximum ratio between two row norms:
denote the maximum ratio between two row norms: ![\[\gamma \coloneqq \frac{ \max_i \norm{a_i}}{\min_i \norm{a_i}}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9289f9e9a0f729efb516fbda78ac3453_l3.png "Rendered by QuickLaTeX.com")
![\[\kappa_{\rm dem}(A) \le \gamma \cdot \kappa_{\rm dem}(D_A A).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d842439abf3ace1c3c590d983ec36107_l3.png "Rendered by QuickLaTeX.com")
 , there exists a matrix
, there exists a matrix  where this bound is nearly attained:
where this bound is nearly attained: ![\[\kappa_{\rm dem}(A_\gamma) \ge \sqrt{1-\frac{1}{n}} \cdot \gamma \cdot \kappa_{\rm dem}(D_{A_\gamma}A_\gamma).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9ff68e369e8f22f56563fdc443894ce4_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{D_AA}_{\rm F} = \sqrt{n}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-dc083ac4d61189b0851e1495ff99f25d_l3.png "Rendered by QuickLaTeX.com")
 as follows
as follows ![\[\frac{1}{\sigma_{\rm min}(D_A A)} = \norm{A^\dagger D_A^{-1}}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c96cc4653bb180133ed40dd1c84687fd_l3.png "Rendered by QuickLaTeX.com")
 denotes the
denotes the  , we have
, we have ![\[\frac{1}{\sigma_{\rm min}(D_A A)} = \norm{A^\dagger D^{-1} (DD_A^{-1})} \le \norm{A^\dagger D^{-1}} \norm{DD_A^{-1}} = \frac{\norm{DD_A^{-1}}}{\sigma_{\rm min}(DA)}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a65986b5ef3ac5975280bffcd2fcb927_l3.png "Rendered by QuickLaTeX.com")
 is diagonal its spectral norm is
is diagonal its spectral norm is ![\[\norm{DD_A^{-1}} = \max \left\{ \frac{|D_{ii}|}{|(D_A)_{ii}|} : 1\le i \le n \right\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-705ca33a5372c0bdf858770f9d204436_l3.png "Rendered by QuickLaTeX.com")
 are
are  , so
, so ![\[\norm{DD_A^{-1}} = \max \left\{ |D_{ii}|\norm{a_i} : 1\le i\le n \right\}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b8026880c5418d6aac0894b839079753_l3.png "Rendered by QuickLaTeX.com")
 . The maximum row norm is always less than the largest singular value of
. The maximum row norm is always less than the largest singular value of  . Therefore, combining this result, (7), and (9), we obtain
. Therefore, combining this result, (7), and (9), we obtain ![\[\kappa_{\rm dem}(D_AA) \le \sqrt{n} \cdot \frac{\sigma_{\rm max}(DA)}{\sigma_{\rm min}(DA)} = \sqrt{n}\cdot \kappa (DA).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9e73680b6f4fe21817d43adb57a457ff_l3.png "Rendered by QuickLaTeX.com")
 , we are free to minimize over
, we are free to minimize over ![\[\kappa_{\rm dem}(D_AA) \le \sqrt{n}\cdot \min_{D \in \mathrm{Diag}} \kappa (DA).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e18d74284219efc70ced6833db98a733_l3.png "Rendered by QuickLaTeX.com")
 . Using the Moore–Penrose pseudoinverse again, write
. Using the Moore–Penrose pseudoinverse again, write ![\[\kappa_{\rm dem}(A) = \norm{D_A^{-1}(D_AA)}_{\rm F} \norm{(D_A A)^\dagger D_A}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-50cf827241cc20f905230e4a51946d0b_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{BC}_{\rm F} \le \norm{B}\norm{C}_{\rm F}, \quad \norm{BC} \le\norm{B}\norm{C}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-051f8f43c351dcc02a4d3342d55b5455_l3.png "Rendered by QuickLaTeX.com")
![\[\kappa_{\rm dem}(A) \le \norm{D_A^{-1}}\norm{D_AA}_{\rm F} \norm{(D_A A)^\dagger} \norm{D_A}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1d3aa06275558feecad7fe4a38c3f64a_l3.png "Rendered by QuickLaTeX.com")
 and
and  . We conclude
. We conclude  . Then
. Then  with
with  and
and ![\[\kappa_{\rm dem}(A_\gamma) = \frac{\norm{A_{\gamma}}_{\rm F}}{\sigma_{\rm min}(A_\gamma)} = \frac{\sqrt{(n-1)\gamma^2+1}}{1} \ge \sqrt{n} \cdot \sqrt{1-\frac{1}{n}} \cdot \gamma.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0216b29d23ab394e7040423bd9a13ebe_l3.png "Rendered by QuickLaTeX.com")
 more iterations than standard RK on a worst-case example, which can be a big difference for large problems. But, particularly for badly row-scaled problems, Proposition 2 shows that uniform RK can dramatically outcompete standard RK. Ultimately, I would give two answers.
more iterations than standard RK on a worst-case example, which can be a big difference for large problems. But, particularly for badly row-scaled problems, Proposition 2 shows that uniform RK can dramatically outcompete standard RK. Ultimately, I would give two answers.![\[W \gets W - \eta G,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f4e1c8e417c22f5378f2c084fe8937cd_l3.png "Rendered by QuickLaTeX.com")
![\[W\gets W - \eta \operatorname{polar}(G).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c9c2f2804d2675c9a8a9e6112b43c6f1_l3.png "Rendered by QuickLaTeX.com")
![\[\operatorname{polar}(G) \coloneqq \operatorname*{argmin}_{Q \textrm{ with orthonormal columns}} \norm{G - Q}_{\rm F}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-cedee2dc450e00d845f4b3f41de98a5f_l3.png "Rendered by QuickLaTeX.com")
 .
.
 , the polar factor may be computed in closed form as
, the polar factor may be computed in closed form as  . But computing the SVD is computationally expensive, particularly in GPU computing environments. Are there more efficient algorithms that avoid the SVD? In particular, can we design algorithms that use only matrix multiplications, for maximum GPU efficiency?
. But computing the SVD is computationally expensive, particularly in GPU computing environments. Are there more efficient algorithms that avoid the SVD? In particular, can we design algorithms that use only matrix multiplications, for maximum GPU efficiency?![f[G] \coloneqq Uf(\Sigma)V^\top](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ced13a661f24d5413c3535383f4c910f_l3.png "Rendered by QuickLaTeX.com") .
. is not odd. But, to obtain the polar factor, we only need a function
is not odd. But, to obtain the polar factor, we only need a function ![\[\operatorname{sign}(x) = \begin{cases} 1, & x > 0, \\ 0, & x = 0, \\ -1, & x < 0. \end{cases}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-103d0549df7eab731cd55caeaef8f0f2_l3.png "Rendered by QuickLaTeX.com")
![\[\operatorname{polar}(G) = \operatorname{sign}[G].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-94977973d95c7d3483bee0620f2ef5f9_l3.png "Rendered by QuickLaTeX.com")
 , we have
, we have ![\[p[G] = a_1 G + a_3 G(G^\top G) + \cdots + a_{2k+1} G(G^\top G)^k.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3377aab54a08cc6e010a9df4e06d0606_l3.png "Rendered by QuickLaTeX.com")
![f[G]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a72a56b33f34eecf4bf37ad1ee1c78ea_l3.png "Rendered by QuickLaTeX.com") by first approximating
by first approximating ![p[G]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2f3968e6285f4d4baef540b858d2c50a_l3.png "Rendered by QuickLaTeX.com") as a proxy for
as a proxy for ![\sin[G]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7d43149d58358ce19f9f50e096f525e2_l3.png "Rendered by QuickLaTeX.com") using its degree-three Taylor polynomial.
using its degree-three Taylor polynomial. . However, this approach converges fairly slowly as we increase the degree
. However, this approach converges fairly slowly as we increase the degree  is a fixed point of the
is a fixed point of the  function:
function: ![\[\operatorname{sign}(\operatorname{sign}(x)) = \operatorname{sign}(x) \quad \text{for all } x \in \real.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-fa3bb06693b0d6b4742c882a7a2f4b7c_l3.png "Rendered by QuickLaTeX.com")
 applying the following fixed point equation until convergence:
applying the following fixed point equation until convergence: ![\[P \gets \frac{3}{2} P - \frac{1}{2} PP^\top P.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-88631b351c7b5aafba34c5d29a5c4921_l3.png "Rendered by QuickLaTeX.com")
 , resulting in an approximation of the form
, resulting in an approximation of the form ![\[\operatorname{sign}[G] \approx p_t[p_{t-1}[\cdots[p_2[p_1[G]]\cdots]].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-fa6a1907a8c5265dd61a630d5f0eb268_l3.png "Rendered by QuickLaTeX.com")
 ?
? seemly could depend in a complicated way on all of the previous polynomials
seemly could depend in a complicated way on all of the previous polynomials  . Fortunately, the authors of The Polar Express show that there is a very simple way of computing the optimal polynomials. Begin by assuming that the singular values of
. Fortunately, the authors of The Polar Express show that there is a very simple way of computing the optimal polynomials. Begin by assuming that the singular values of ![[\ell_0,u_0]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-211f4ff3af7863232d7db347c4ef7e54_l3.png "Rendered by QuickLaTeX.com") . We then choose
. We then choose  to be the degree-(
to be the degree-( error:
error:![\[p_1 = \operatorname*{argmin}_{\text{odd degree-($2k+1$) polynomial } p} \max_{x \in [\ell_0,u_0]} |p(x) - \operatorname{sign}(x)|.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e291c7f68e60512be5e8e92c7e3cf90b_l3.png "Rendered by QuickLaTeX.com")
![p_1[G]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-630611cd5d04a5cd2fabb16f5855dcfa_l3.png "Rendered by QuickLaTeX.com") lie in a new interval
lie in a new interval ![[\ell_1,u_1]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-15fd6cfaa78a33099efd2e7ebe256fbb_l3.png "Rendered by QuickLaTeX.com") . To build the next polynomial
. To build the next polynomial  , we simply find the optimal approximation to the sign function on this interval:
, we simply find the optimal approximation to the sign function on this interval: ![\[p_2 = \operatorname*{argmin}_{\text{odd degree-($2k+1$) polynomial } p} \max_{x \in [\ell_1,u_1]} |p(x) - \operatorname{sign}(x)|.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d4b8a6d502f41db5153e14b3a0f17b88_l3.png "Rendered by QuickLaTeX.com")
 as we want.
as we want.
 and
and  , the coefficients of the optimal polynomials
, the coefficients of the optimal polynomials  by normalizing
by normalizing  . As such, there is only one parameter
. As such, there is only one parameter  is appropriate. (As the authors stress, their method remains convergent even if too large a value of
is appropriate. (As the authors stress, their method remains convergent even if too large a value of ![\[\chi^2\left(\rho^{(n)} \, \middle|\middle| \, \pi\right) \le \left( \max \{ \lambda_2, -\lambda_n \} \right)^{2n} \chi^2\left(\rho^{(0)} \, \middle|\middle| \, \pi\right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4083a8792e3728ec282c234e8714a3e0_l3.png "Rendered by QuickLaTeX.com")
 denotes the distribution of the chain at time
denotes the distribution of the chain at time  denotes the
denotes the  denotes the
denotes the 
 denote the decreasingly ordered eigenvalues of the Markov transition matrix
denote the decreasingly ordered eigenvalues of the Markov transition matrix  .
.
 . In this post, we will talk about techniques for bounding
. In this post, we will talk about techniques for bounding  and functions
and functions  , treating them as one and the same
, treating them as one and the same  .
.![\expect_\pi[f]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5bf080ed72a8c1f4965ae7500b95d55e_l3.png "Rendered by QuickLaTeX.com") and
and  denote the variance with respect to the stationary distribution
denote the variance with respect to the stationary distribution ![\[\expect_\pi[f] = \sum_{i=1}^m f(i) \pi_i, \quad \Var_\pi(f) \coloneqq \expect_\pi[(f-\expect_\pi[f])^2].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-140e8451d7640ea23691bc6e0ac79296_l3.png "Rendered by QuickLaTeX.com")
![\[\langle f, g\rangle \coloneqq \expect_\pi[f\cdot g] = \sum_{i=1}^m f(i) g(i) \pi_i.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-429786c5a31567611cd2b34a899b00c7_l3.png "Rendered by QuickLaTeX.com")
![\expect_{x \sim \sigma, y\sim \tau} [f(x,y)]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c2a42f5e811f88ddfb5da1a1164bd034_l3.png "Rendered by QuickLaTeX.com") to denote the expectation of
to denote the expectation of  where
where  and
and  .
.
 are orthonormal in the
are orthonormal in the ![\[\langle \varphi_i ,\varphi_j\rangle = \begin{cases}1, & i = j, \\0, & i \ne j.\end{cases}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1bcdaefcfb18b68324fe7793e2cb3534_l3.png "Rendered by QuickLaTeX.com")
 from its mean, where
from its mean, where ![\[\Var_\pi(f) = \frac{1}{2} \expect_{x,y \sim \pi} [(f(x) - f(y))^2].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-63885f39b045d90e13763469722ab480_l3.png "Rendered by QuickLaTeX.com")
 .
. be sampled from the stationary distribution, and let
be sampled from the stationary distribution, and let  denote one step of the Markov chain after
denote one step of the Markov chain after  . The local variance is
. The local variance is![\[\mathcal{E}(f) = \frac{1}{2} \expect [(f(x_0) - f(x_1))^2].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a1f334058762ab02c87e271c2c92bc47_l3.png "Rendered by QuickLaTeX.com")
 if
if ![\[\Var_\pi(f)\le \alpha \cdot \mathcal{E}(f) \quad \text{for every function } f.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5e8793ac1bfab318d0a5010d48b2dbcf_l3.png "Rendered by QuickLaTeX.com")
 is close to
is close to  ,
,  , etc.; the function
, etc.; the function ![\[\alpha= \frac{1}{1-\lambda_2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0143e8947cd9b6ad343afca4ab36be9f_l3.png "Rendered by QuickLaTeX.com")
![\[\lambda_2 \le \frac{1}{1-\alpha}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-be80ee6ab2085c10ff126a991ec9ca19_l3.png "Rendered by QuickLaTeX.com")
 that have finitely many possible states and are indexed by discrete times
that have finitely many possible states and are indexed by discrete times  .. We can generalize Markov chains by lifting both of these restrictions, considering Markov processes
.. We can generalize Markov chains by lifting both of these restrictions, considering Markov processes  which take values in continuous space (such as the real line
which take values in continuous space (such as the real line  ) and are indexed by continuous times
) and are indexed by continuous times  .
.
![\[x_t = e^{-t}x_0 + e^{-t} B_{e^{2t}-1},\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c98a0f6993431b8ea3c5ce14e96fd6d0_l3.png "Rendered by QuickLaTeX.com")
 denotes a (standard)
denotes a (standard)  has a
has a  is has a Gaussian distribution with mean
is has a Gaussian distribution with mean  and variance
and variance  .
. denote a standard Gaussian random variable, Gaussian Poincaré inequality states that
denote a standard Gaussian random variable, Gaussian Poincaré inequality states that![\[\Var(f(Z)) \le \expect \big[(f'(Z))^2\big].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2da5301cf15bd60eddb662b9859d986b_l3.png "Rendered by QuickLaTeX.com")
![\[\mathcal{E}(f) = \expect \big[(f'(Z))^2\big].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c734423684e8cbeef463d21fd3a7d8cd_l3.png "Rendered by QuickLaTeX.com")
 is controlled by its local variability, here quantified by the expected squared derivative:
is controlled by its local variability, here quantified by the expected squared derivative: as a generalization of the “expected squared derivative” of the function
as a generalization of the “expected squared derivative” of the function  has derivative bounded by
has derivative bounded by  . Thus,
. Thus,![\[\Var(\tanh Z) \le \expect[(f'(Z))^2] \le 1.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-62932f1a7dd99680d5022f2f617204f6_l3.png "Rendered by QuickLaTeX.com")
 is
is ![\Var(\tanh(Z)) = 0.5\expect[(\tanh Z - \tanh Z')^2] \le 0.5 \expect[(Z-Z')^2] = \Var(Z) = 1](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-13cfee974435cfa789dc50791f5dd980_l3.png "Rendered by QuickLaTeX.com") , where
, where  is an independent copy of
is an independent copy of ![\[\Var_\pi(f)\le \frac{1}{1-\lambda_2}\cdot\mathcal{E}(f) \quad \text{for all } f\in\real^m.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8b1dc6392929fce027a5637445e5e788_l3.png "Rendered by QuickLaTeX.com")
![\expect[f] = 0](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ec4690eff3c2fed1a114233e551241f6_l3.png "Rendered by QuickLaTeX.com") to prove (3). Next, we derive formulas for
to prove (3). Next, we derive formulas for  . We conclude by expanding
. We conclude by expanding ![\expect_\pi[f]=0](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6fa817f7b45e4b28fb57943a37ce1887_l3.png "Rendered by QuickLaTeX.com") . Indeed, both the variance
. Indeed, both the variance  denote the function
denote the function![\[\mathbb{1}(i) = 1 \quad\text{for }i =1,\ldots,m,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-76d08dac199d8cde7c208717c678cb21_l3.png "Rendered by QuickLaTeX.com")
![\[\Var_\pi(f+c\mathbb{1})=\Var_\pi(f)\quad\text{and}\quad\mathcal{E}(f+c\mathbb{1})=\mathcal{E}(f)\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-397546ace1cf7f9ee61e6b903c5d30fa_l3.png "Rendered by QuickLaTeX.com")
![\[\expect_\pi[f] = 0.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-768127e484e8e86a1ea3b0c57ff2254b_l3.png "Rendered by QuickLaTeX.com")
![\expect_\pi[f] = 0](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5bb3a81c46de080d7497a8b15dc671e4_l3.png "Rendered by QuickLaTeX.com") . Then the variance is
. Then the variance is![\[\Var_\pi(f)=\expect[f^2]=\sum_{i=1}^m f(i)f(i)\pi_i.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-34c04d78b686f1464575345ff7942f0e_l3.png "Rendered by QuickLaTeX.com")
![\[\Var_\pi(f)=\langle f,f\rangle.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c8e6cda6b300b5ffc2d56c82e0024a40_l3.png "Rendered by QuickLaTeX.com")
![\[\mathcal{E}(f) = \frac{1}{2} \expect[(f(x_0)-f(x_1))^2]\quad \text{where }x_0\sim\pi.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b19838fb6f36da7cbd443083ba89017c_l3.png "Rendered by QuickLaTeX.com")
 and
and  is
is  . Thus,
. Thus,![\[\mathcal{E}(f) = \frac{1}{2} \sum_{i,j=1}^m (f(i)-f(j))^2 \pi_iP_{ij}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3b001418aee305354517a3784887e27f_l3.png "Rendered by QuickLaTeX.com")
![\[\mathcal{E}(f) = {\rm A} + {\rm B} + {\rm C}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a50f3c027ad34072a0bd9bd6348c40a0_l3.png "Rendered by QuickLaTeX.com")

 , recognize that
, recognize that  . Thus,
. Thus, ![\[{\rm A} = \frac{1}{2}\sum_{i=1}^m (f(i))^2 \pi_i = \frac{1}{2}\langle f, f\rangle.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-dad7cb3fe33d90897dd8b5a31625ca44_l3.png "Rendered by QuickLaTeX.com")
 , use detailed balance
, use detailed balance  . Then, using the condition
. Then, using the condition  , we obtain
, we obtain![\[{\rm B} = \frac{1}{2} \sum_{j=1}^m (f(j))^2 \left(\sum_{i=1}^m \pi_j P_{ji} \right) = \frac{1}{2} \sum_{j=1}^m (f(j))^2 \pi_j = \frac{1}{2} \langle f, f\rangle.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d26f896a5e016a55ba59c4b0abca8f69_l3.png "Rendered by QuickLaTeX.com")
 , recognize that
, recognize that  is the
is the  . Thus,
. Thus,![\[{\rm C} = - \sum_{i=1}^m f(i) Pf(i) \,\pi_i = -\langle f, Pf\rangle.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-37f0ed03af3cbfc11bccf5a6f6e20bb1_l3.png "Rendered by QuickLaTeX.com")
![\[\mathcal{E}(f) = \langle f, (I-P)f\rangle,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9450ee3a751ddf403b01a7f69fe26a2e_l3.png "Rendered by QuickLaTeX.com")
 denotes the identity matrix.
denotes the identity matrix.
![\[\Var_\pi(f) \le \frac{1}{1-\lambda_2} \cdot\mathcal{E}(f) \quad \text{for all $f$ with $\expect_\pi[f] = 0$}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3d732320f8d8787a3cc4cec15b09d8d4_l3.png "Rendered by QuickLaTeX.com")
![\[\frac{\mathcal{E}(f)}{\Var_\pi(f)}\ge 1-\lambda_2 \quad \text{for all $f$ with $\expect_\pi[f] = 0$}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f7218fec7abdef7e7270acb4dbeb6fd7_l3.png "Rendered by QuickLaTeX.com")
![\[\frac{\langle f, (I-P)f\rangle}{\langle f, f\rangle} \ge 1-\lambda_2 \quad \text{for all $f$ with $\expect_\pi[f] = 0$}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3690797d7d53191ee73aac81fbcdd36c_l3.png "Rendered by QuickLaTeX.com")
![\[f = c_1 \varphi_1 + c_2 \varphi_2 + \cdots + c_m\varphi_m.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-df5f258e09e7a807979852d3373b2035_l3.png "Rendered by QuickLaTeX.com")
 .
.
 , we have that
, we have that

![\[a_i = \frac{c_i^2}{c_2^2 + \cdots + c_m^2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3895b1bbfe12ec753d65b2a519517c7b_l3.png "Rendered by QuickLaTeX.com")
 are nonnegative and add to
are nonnegative and add to ![\[a_2+\cdots+a_m = \frac{c_2^2+\cdots+c_m^2}{c_2^2+\cdots+c_m^2} = 1.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b076fc822731dc667d48e67a60bbf6ea_l3.png "Rendered by QuickLaTeX.com")
 and
and  (equivalently, setting
(equivalently, setting  ). Thus, we conclude
). Thus, we conclude![\[\frac{\langle f, (I-P)f\rangle}{\langle f, f\rangle}\ge 1-\lambda_2,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1b798877b91df792cf96ef10eb9ac632_l3.png "Rendered by QuickLaTeX.com")
 .
.
 of two
of two  is positive semidefinite (psd, for short) if it is
is positive semidefinite (psd, for short) if it is  for all vectors
for all vectors  . All matrices in this post are real, though the proofs we’ll consider also extend to complex matrices. The entrywise product will be denoted
. All matrices in this post are real, though the proofs we’ll consider also extend to complex matrices. The entrywise product will be denoted  and is defined as
and is defined as  . The entrywise product is also known as the Hadamard product or Schur product.
. The entrywise product is also known as the Hadamard product or Schur product. :
: ![\[x^\top (A\circ M)x = \sum_{i,j=1}^n x_i (A\circ M)_{ij} x_j = \sum_{i,j=1}^n x_i A_{ij} M_{ij} x_j.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ad02e17a6c90aca9b469ef0fe73bb1d3_l3.png "Rendered by QuickLaTeX.com")
![\[x^\top (A\circ M)x = \sum_{i,j=1}^n x_i A_{ij} x_j M_{ji} = \tr(\operatorname{diag}(x) A \operatorname{diag}(x) M).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4386c5381ce8bcc4e8ded09b2c00bcf5_l3.png "Rendered by QuickLaTeX.com")
 ). Thus, we may write
). Thus, we may write  . Substituting these expressions in the trace formula and invoking the
. Substituting these expressions in the trace formula and invoking the ![\[x^\top (A\circ M)x = \tr(\operatorname{diag}(x) B^\top B \operatorname{diag}(x) C^\top C) = \tr(C\operatorname{diag}(x) B^\top B \operatorname{diag}(x) C^\top).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5f330914f0a1aa93654b4330e2615c7b_l3.png "Rendered by QuickLaTeX.com")
![\[C\operatorname{diag}(x) B^\top B \operatorname{diag}(x) C^\top = G^\top G \quad \text{for } G = B \operatorname{diag}(x) C^\top.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-27588c5bd3897e90305feba5e03a6488_l3.png "Rendered by QuickLaTeX.com")
![\[x^\top (A\circ M)x = \tr(G^\top G) \ge 0.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c7e68b9a2fc38f723e37e208c2d4c28b_l3.png "Rendered by QuickLaTeX.com")
 for every vector
for every vector  and
and  denote the
denote the ![\[A = \sum_i b_ib_i^\top \quad \text{and} \quad M = \sum_j c_jc_j^\top.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-95d0f789d3f2d56faa06e53e7bc33366_l3.png "Rendered by QuickLaTeX.com")
![\[A\circ M = \sum_{i,j} (b_ib_i^\top \circ c_jc_j^\top).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c808a8bf106990528aae134391107813_l3.png "Rendered by QuickLaTeX.com")
 and
and  is, by direct computation,
is, by direct computation,  . Thus,
. Thus, ![\[A\circ M = \sum_{i,j} (b_i\circ c_j)(b_i\circ c_j)^\top\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5484fa9e5570cb1c2a639512b48c303c_l3.png "Rendered by QuickLaTeX.com")
 is seen to have zero mean as well. Thus, the
is seen to have zero mean as well. Thus, the  entry of the covariance matrix
entry of the covariance matrix  of
of ![\[\expect[x_iy_ix_jy_j] = \expect[x_ix_j] \expect[y_iy_j] = A_{ij} M_{ij} = (A\circ M)_{ij}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b792b112f5388bf21a7f6254af9b271d_l3.png "Rendered by QuickLaTeX.com")
 have replaced by expectations
have replaced by expectations ![A = \expect [xx^\top]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ec158da95565c147435a98aff8fdd42c_l3.png "Rendered by QuickLaTeX.com") .
. of two psd matrices
of two psd matrices ![\[A\circ M = ((A\otimes M)_{(i+n(i-1))(i+n(i-1))} : i = 1,\ldots,n).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-46b3a14143b4f365e5cd92118733a1bb_l3.png "Rendered by QuickLaTeX.com")